
特立氟胺微乳的制备及其体外透皮吸收研究
2018-11-06 09:33曹雅茹程节玲颜传文
安徽医科大学学报 2018年10期
王 东,曹雅茹,金 涌,程节玲,颜传文
特立氟胺(teriflunomide, TEF) 是类风湿关节炎治疗药物来氟米特(leflunomide, LEF)在体内的活性代谢物。LEF通过体内生成TEF而抑制二氢乳酸脱氢酶(DHODH),阻碍来自淋巴细胞的嘧啶生成,干扰酪氨酸脱氢酶的活性。LEF由于其严重的肝损害,2010年美国FDA予以黑框警告。2012年特立氟胺口服制剂Aubagio在美国上市,上市后患者因无法耐受胃肠道反应和肝毒性而停止服药。FDA已对Aubagio致严重肝损伤风险予黑框警告。因此如何能减轻或避免TEF肝损害的研究具有重要意义。微乳 (microemulsion, ME)是由四相(油相、水相、乳化剂和助乳化剂)组成的,具有澄清透明、黏度低、热力学稳定和各向同性等特点的油水混合体系。其粒径介于10~100 nm,制备程序简单,操作方便,无需特殊设备,可通过过滤除菌[1-3]。外用ME体内可通过淋巴转运后吸收,能减少胃肠道代谢过程及肝脏的首过效应,减轻肝毒性[4]。该研究组通过对特立氟胺ME的处方筛选、优化,制备最佳处方比的特立氟胺外用ME,并对其体外透皮性能进行考察,为后续特立氟胺ME体内药代动力学研究做铺垫。
1 材料与方法
1.1仪器Franz扩散池(自制);LC-20 A 型高效液相色谱仪(日本岛津公司);NanoZS 90型激光纳米粒度仪(英国马尔文公司);79 HW-1型恒温磁力搅拌器(江苏金坛金城国胜实验仪器厂);JEM-100 SX型透射式电子显微镜(日本电子株式会社);DDS-11 AW 型电导率仪(上海般特仪器有限公司);Milli-Q Integral 15型超纯水机(美国Millipore公司)。
1.2试剂特立氟胺对照品(自制,质量分数为99%);辛癸酸甘油酯(ODO,山东西亚试剂公司,批号:T0809);大豆磷脂(Lecithin,上海艾韦特有限公司,批号:170103);油酸(OA,阿拉丁试剂公司,批号:160129);肉豆蔻酸异丙酯(IPM,国药集团化学试剂,批号:17A0305);吐温80(天津光复精细化工研究所,批号:20170301);水为双蒸水;其余试剂均为分析纯。
1.3动物SD大鼠,雄性,8只,普通级,体重(225±25)g,由安徽医科大学实验动物中心提供。给予正常饮食,在标准环境饲养1周。
1.4方法
1.4.1ME伪三元相图的绘制 按一定质量比(Km=乳化剂/助乳化剂),分别精密称取乳化剂和助乳化剂于透明、干燥的具塞安瓿瓶中,相互混合并溶解,即得混合乳化剂,将油相与混合乳化剂按如下质量比(9 ∶1、8 ∶2、7 ∶3、6 ∶4、5 ∶5、4 ∶6、3 ∶7、2 ∶8、1 ∶9 )分别混合,恒温磁力搅拌器搅拌,观察浑浊情况,在澄明液中缓慢滴加双蒸水,肉眼观察液体由澄明转为浑浊(即临界点),记录此时ME组成的质量百分比。采用Origin8.0软件对伪三元相图的相关数据进行处理,并绘制伪三元相图,比较ME区域大小。
1.4.2组分互溶性考察及助乳化剂的选择 选择司盘80、大豆磷脂、司盘80-吐温80复合物(MIX 80,HLB=5.0)为乳化剂,异丙醇、无水乙醇、1,2-丙二醇、PEG 400为助乳化剂。观察25 ℃下不同乳化剂与助乳化剂的互溶情况(溶解性、流动性、澄明度),初步确定混合乳化剂的组成。
1.4.3处方筛选
1.4.3.1乳化剂的选择 25 ℃下,以无水乙醇为助乳化剂,IPM为油相,固定Km值=1 ∶1,分别考察大豆磷脂、司盘80、MIX 80所形成的ME区域大小。
1.4.3.2油相的选择 25 ℃下,固定Km值(乳化剂/助乳化剂)为1 ∶1,油相与混合乳化剂质量比6 ∶4,比较油相为OA、IPM、ODO、EO时ME区域大小。
1.4.3.3混合乳化剂比例的确定 25 ℃下,以油相与混合乳化剂质量比1 ∶1,考察Km值为1 ∶2、2 ∶3、1 ∶1、3 ∶2和2 ∶1时ME区域面积大小。
1.4.4ME的制备 将乳化剂与助乳化剂混合溶解,再加入油相混合,在搅拌下将特立氟胺水溶液缓慢滴入上述液体中,搅拌30 min,即得特立氟胺ME。
1.4.5ME质量评价方法
1.4.5.1性状 目视观察制备的特立氟胺ME的性状。
1.4.5.2特立氟胺ME的鉴别 ① 染色法:取A、B两烧杯,加入相同体积特立氟胺ME,A加2滴亚甲蓝(水性染料),B加2滴苏丹红(油性染料),室温静置10 min; ② 电导率法: 取同体积特立氟胺ME、空白ME、特立氟胺水溶液和IPM。采用电导率仪测定样品电导率。
1.4.5.3ME的粒径分布 取“1.4.4”制备的ME,用粒度仪测定ME的粒径分布。
1.4.5.4ME显微形态的观察 常温下取适量稀释5倍的特立氟胺ME滴在铜网上(覆有支持膜),静置15 min,待自然晾干后用1%磷钨酸溶液负染10 min,自然挥干,透射电镜下观察。
1.4.5.5稳定性考察 取特立氟胺ME适量在3 000 r/min离心10 min,结果ME仍澄清、透明,且不分层。另取特立氟胺ME 20 ml于具塞试管中,在室温条件下放置6个月,分别于0、1、2、3、6个月观察体系是否透明、澄清,有无分层及沉淀等现象,并测定ME中药物含量、电导率。
1.4.6含量测定方法
1.4.6.1溶液的配制 ① 对照品溶液的配制:精密称取特立氟胺对照品25 mg,加适量甲醇溶解,移入25 ml容量瓶后加甲醇定容摇匀,精密量取5 ml于25 ml容量瓶中,加甲醇定容摇匀,即得200.00 μg/ml的特立氟胺对照品储备液; ② 供试品溶液的配制:精密称取制备的ME适量,加适量甲醇破乳,移入50 ml容量瓶中加甲醇定容后超声30 min,3 000 r/min离心10 min,精密量取10 ml于25 ml容量瓶中,加甲醇定容,微孔滤膜(0.45 μm)滤过得200.00 μg/ml供试品储备液; ③ 空白供试品溶液的配制:将精密称取空白ME 10.00 g,余操作同②。所有配制溶液置4 ℃冰箱保存。
1.4.6.2色谱条件 色谱柱:ODS-C18色谱柱(250 mm×4.6 mm,5 μm),流动相:0.01 mol/L磷酸盐缓冲液(pH 7.4) ∶甲醇=43.8 ∶56.2;流速1.00 ml/min;柱温40 ℃;检测波长294.0 nm;进样量20 μl;内标10.0 μg/ml尼泊金甲酯。
1.4.6.3专属性实验 分别吸取“1.4.6.1”项中的3种溶液,稀释后的按“1.4.6.2”项条件进样。观察空白ME中是否有成分对特立氟胺峰有干扰。
1.4.6.4线性关系考察 精密量取“1.4.6.1”项①储备液,依次用甲醇稀释,得100.00、50.00、25.00、12.50、6.25、3.125 μg/ml的系列标准液,依次进样,以药物峰面积(A)对药物浓度(C, μg/ml)进行线性回归。
1.4.6.5精密度实验 精密量取特立氟胺对照品溶液1 ml于10 ml容量瓶中,加甲醇定容摇匀,按“1.4.6.2”项重复进样(n=6)测定峰面积,根据标准曲线求得浓度,计算精密度。
1.4.6.6稳定性实验 取ME供试品溶液,分别在0、2、4、6、12、24、48 h时进样,按“1.4.6.2”项进样测定峰面积。
1.4.6.7重复性实验 精密量取特立氟胺ME 6份(同批次),按“2.6.1.2”项下制成供试品溶液,按“1.4.6.2”项进样测定峰面积。
1.4.6.8回收率实验 精密量取低、中、高浓度(0.25、1.00、2.50 mg/ml)特立氟胺溶液1 ml,分别加入含空白ME 0.5 g的3个10 ml容量瓶中,分别按“1.4.6.1”项②处理,“1.4.6.2”项平行进样(n=3),计算回收率。
1.4.6.9样品的含量测定 取ME的3个不同批次,按照“1.4.6.1”项②制成溶液,按“1.4.6.2”进样测定。
1.4.7体外透皮实验方法
1.4.7.1鼠皮制备 取大鼠,断颈后刮净背部毛发,剥离背部皮肤(避免损伤),小心去除粘连物和皮下脂肪,用0.9%氯化钠溶液冲洗干净,置0.9%氯化钠溶液中4 ℃保存,7 d内用完。
1.4.7.2样品制备 精密称取ME各成分,按“1.4.4”方法制备特立氟胺ME;采用干胶法制备特立氟胺普通乳剂;另将特立氟胺溶于双蒸水中得特立氟胺水溶液。所有制备的样品避光密封保存。
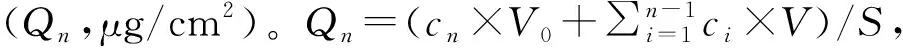
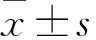
2 结果
2.1ME处方组成与最佳处方比结果
2.1.1组分互溶性和助乳化剂的选择 助乳化剂PEG 400、1,2-丙二醇与各乳化剂的的互溶情况均较差,故不适宜制备ME。不同乳化剂在异丙醇和无水乙醇中均表现出良好的溶解性和流动性,但助乳化剂为异丙醇时体系澄明度差于无水乙醇,故选择无水乙醇为助乳化剂。
2.1.2乳化剂的选择 大豆磷脂所形成ME区域最大,MIX 80和司盘80所形成的ME区域面积均较小。因此确定大豆磷脂为乳化剂。见图1。
2.1.3油相的选择 IPM所形成ME区域最大,EO和ODO的ME区域面积相对较小,OA的ME区域最小。故确定IPM为油相。见图2。
2.1.4混合乳化剂比例的确定 在Km=1 ∶1时,ME区域面积最大,因此确定混合乳化剂Km=1 ∶1。见图3。
2.1.5最佳处方比的确定 该实验水相用量为最大载水量的70%时形成的ME最稳定,结合制备的ME稳定性和成本等因素,最终选择油相与混合乳化剂的质量比为1 ∶1。最终确定最佳处方质量比为IPM ∶大豆磷脂 ∶无水乙醇 ∶双蒸水=42.12 ∶21.06 ∶21.06 ∶15.76。
2.2特立氟胺ME的制备精密称取特立氟胺5.0 mg、IPM 2.106 g、大豆磷脂1.053 g、无水乙醇1.053 g、双蒸水0.783 g,按“1.4.4”制备特立氟胺ME。
2.3特立氟胺ME的质量评价
2.3.1性状 ME为流动性好的黄色澄明液体,存在丁达尔现象。见图4。
2.3.2特立氟胺ME的鉴别
2.3.2.1染色法 苏丹Ⅲ(显红色)在ME中的扩散速度快于亚甲基蓝(显蓝色),故所得ME属油包水(W/O)型。见图5。
2.3.2.2电导率法 实验测得空白ME和特立氟胺ME的电导率值略低于IPM的电导率值,但明显低于特立氟胺水溶液,故判定ME为W/O型ME。
2.3.3ME的粒径分布 粒径分布见图6,测定3次的粒径为(35.18±0.28) nm,PDI为(0.184±0.02),说明ME粒径大小适宜,且粒径范围较窄,分布集中。
2.3.4ME显微形态的观察 电镜照片见图7,在电镜下ME乳滴为表面完整的圆球形,符合ME的粒径要求。
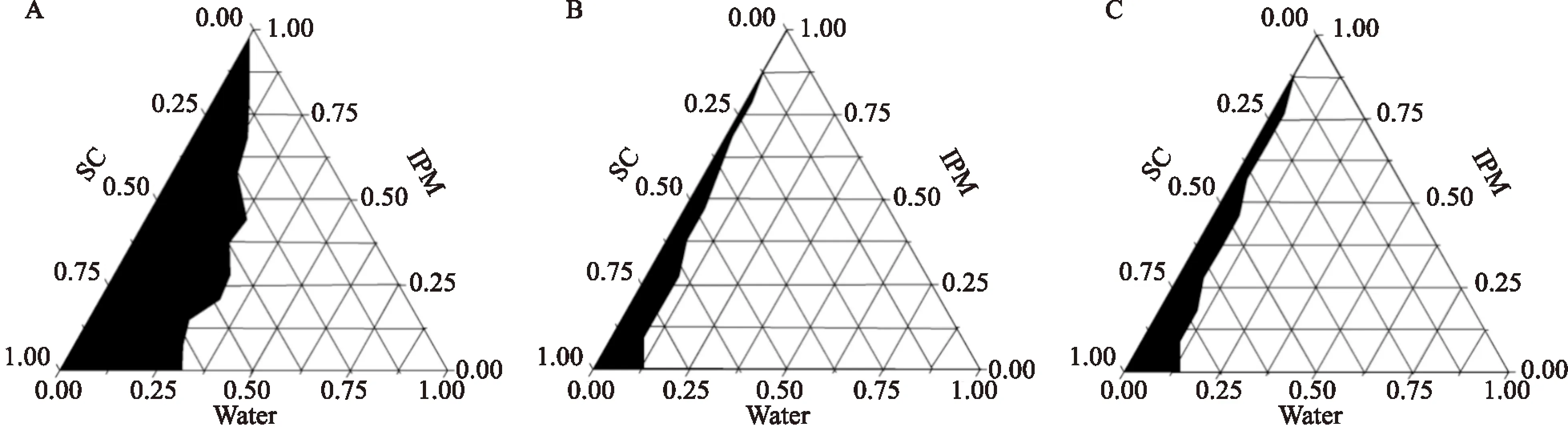
图1 3种不同乳化剂的伪三元相图
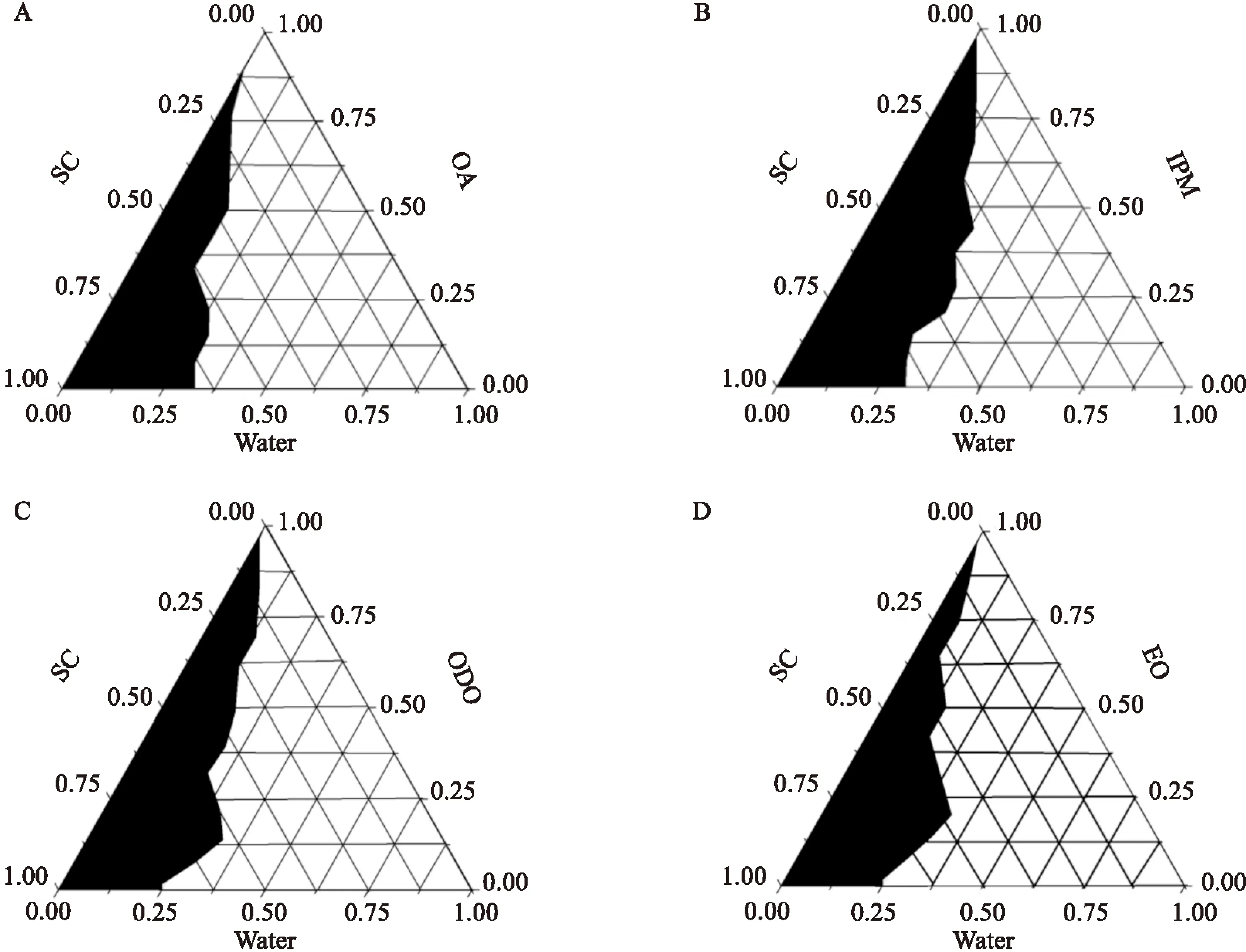
图2 4种油相的伪三元相图
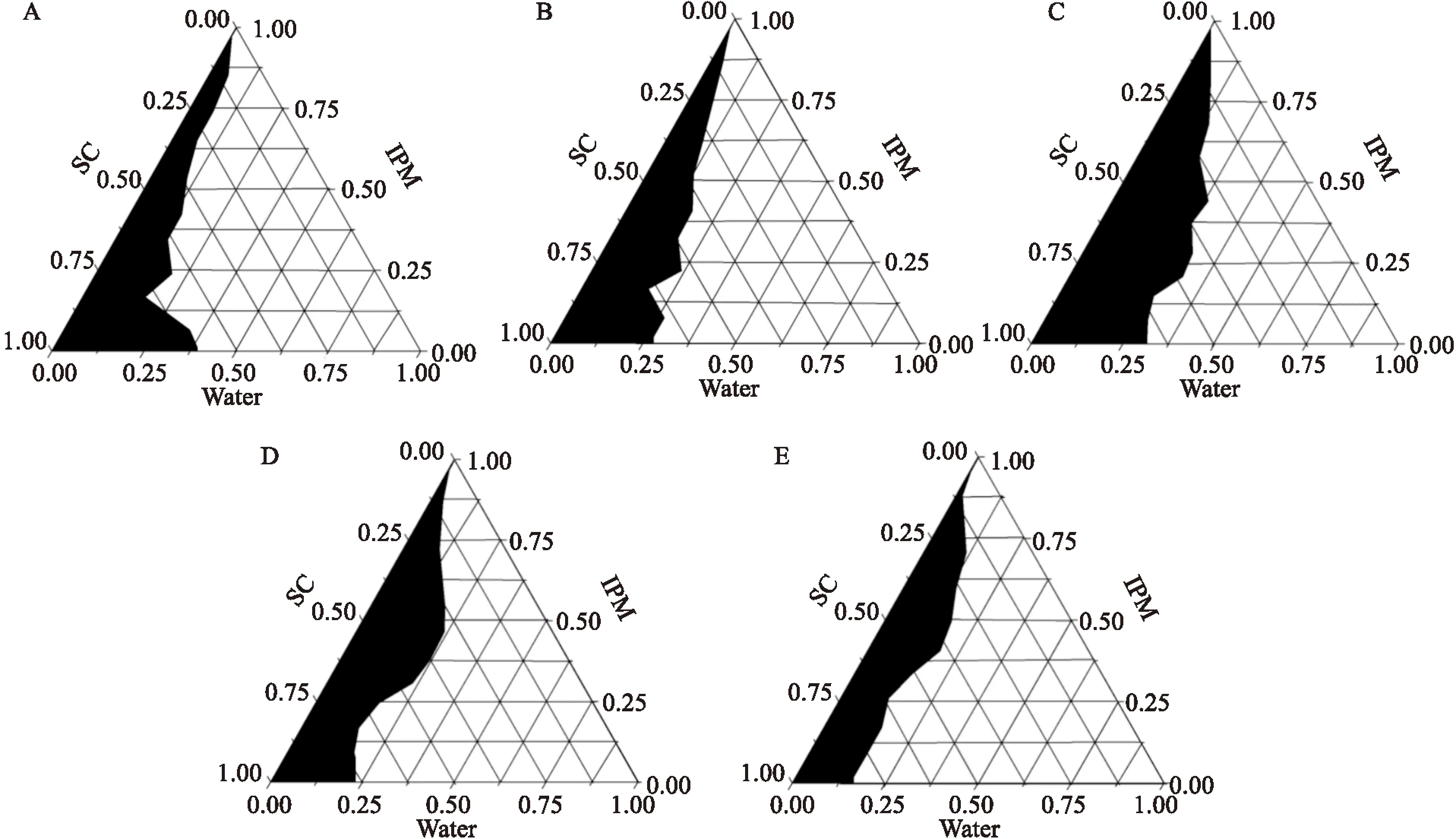
图3 5种Km值的伪三元相图

图4 特立氟胺ME外观

图5 特立氟胺ME类型
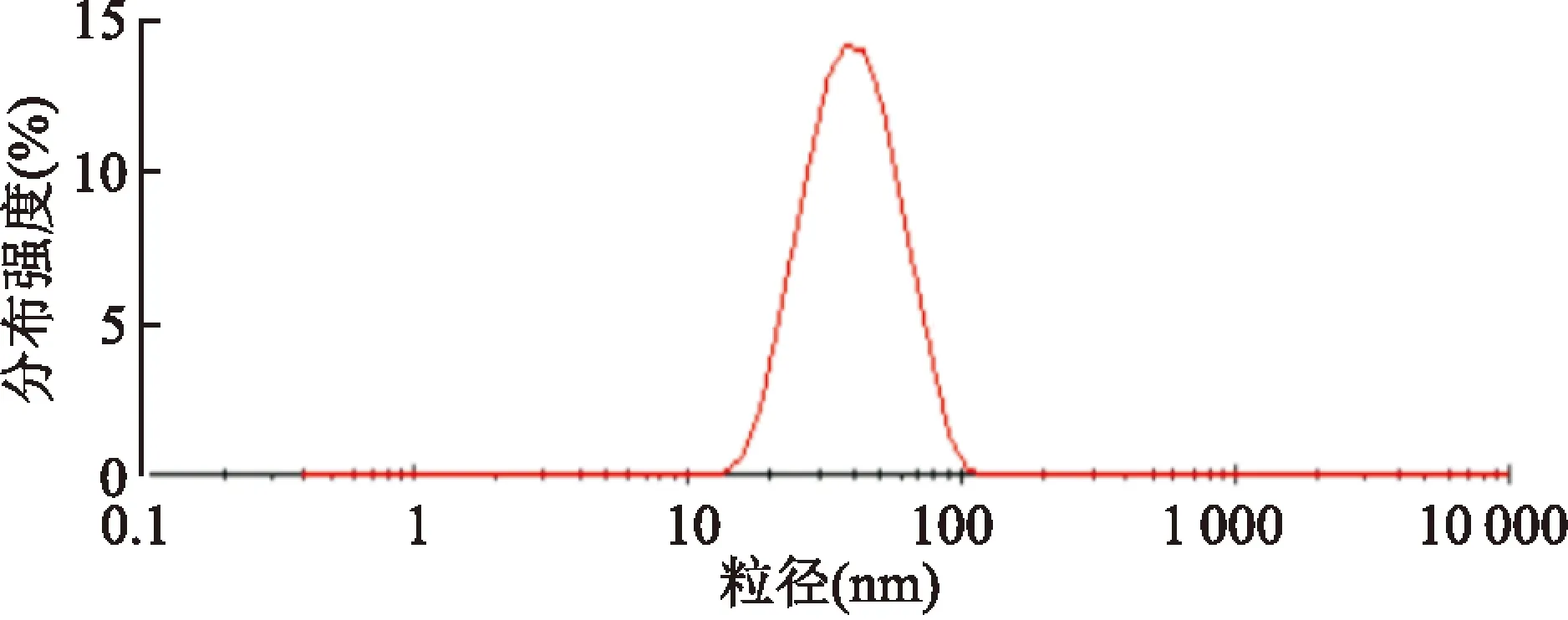
图6 特立氟胺ME的粒径分布
2.3.5稳定性考察 结果体系透明、澄清,无分层及沉淀;药物含量、电导率均保持稳定,说明ME具有良好的稳定性。
2.4特立氟胺ME的含量测定
2.4.1专属性实验 特立氟胺色谱峰不受其他物质干扰,专属性良好,保留时间为10.9 min。见图8。

图7 特立氟胺ME的透射电镜观察 ×124 000
2.4.2线性关系 回归方程为A=0.121 0C-0.512 5(R2=0.999 6),结果显示特立氟胺在3.125~100.00 μg/ml范围内线性良好。
2.4.3精密度实验 结果相对标准偏差(relative standard deviation, RSD)为0.52%,说明仪器精密度良好。
2.4.4稳定性实验 结果RSD为0.79%,结果表明特立氟胺ME稳定性良好。
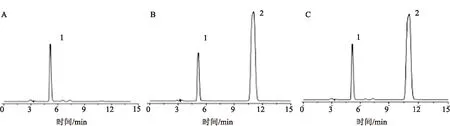
图8 特立氟胺的高效液相色谱图
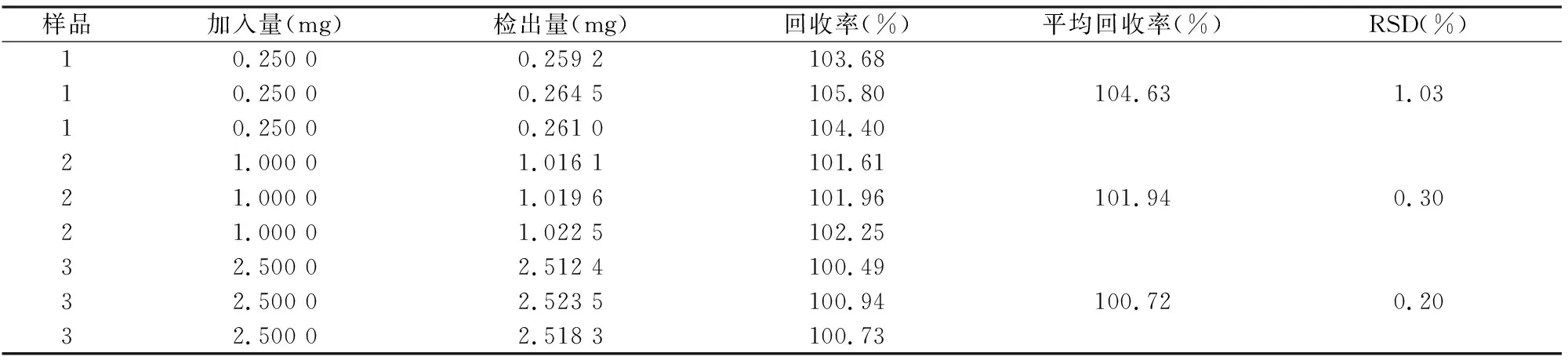
样品加入量(mg)检出量(mg)回收率(%)平均回收率(%)RSD(%)10.250 00.259 2103.6810.250 00.264 5105.80104.631.0310.250 00.261 0104.4021.000 01.016 1101.6121.000 01.019 6101.96101.940.3021.000 01.022 5102.2532.500 02.512 4100.4932.500 02.523 5100.94100.720.2032.500 02.518 3100.73

表2 不同组成特立氟胺制剂
2.4.5重复性实验 计算得峰面积RSD为0.61%,表明该方法重复性良好。
2.4.6回收率实验 结果见表1,3种浓度的回收率均符合要求。
2.4.7样品的含量测定 结果特立氟胺ME含量分别为203.35、194.62、206.09 μg/ml,平均为(201.35±5.99)μg/ml。
2.5体外透皮实验
2.5.1样品制备 按表2精密称取ME各组分,按“1.4.7.2”方法制备,即得表2示的ME、乳剂及水溶液。
2.5.2透皮实验结果 4种特立氟胺样品的渗透曲线见图9。分别以0.1%、0.25%特立氟胺ME的Qn对渗透时间(t)进行回归分析。回归方程分别为Qn=2.698 2t+0.297 1(R2=0.997 1)、Qn=6.138 6t+0.532 3(R2=0.998 1)。0.1%和0.25%特立氟胺ME的Qn对渗透时间(t)线性关系的良好。
4种样品的渗透系数和累计渗透量结果见表3。0.25%特立氟胺ME较0.1%特立氟胺ME的Q24 h显著升高;0.1%特立氟胺ME的Q24 h较其普通乳剂和水溶液亦显著升高,说明ME能有效促进特立氟胺的透皮吸收,且经皮累计渗透量随药物剂量的增加而增多。
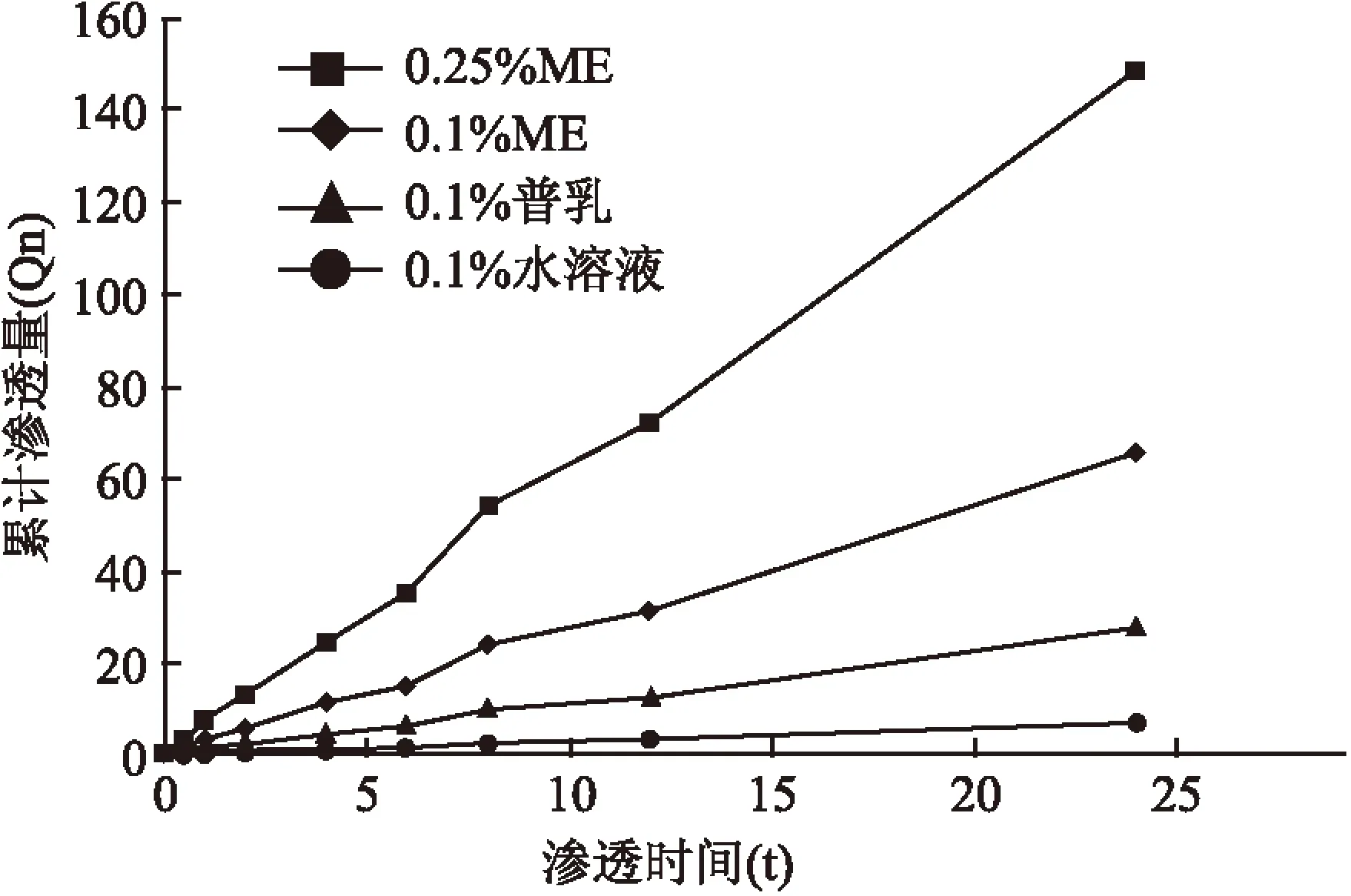
图9 4种特立氟胺样品的渗透曲线图
3 讨论
ME处方各组分的确定及最佳处方比是ME制备研究的重点,亦是难点。在保证ME稳定和最少乳化剂使用的基础上,该研究选择IPM ∶大豆磷脂 ∶无水乙醇 ∶双蒸水比为42.12 ∶21.06 ∶21.06∶15.76,制备了W/O型特立氟胺ME。IPM和ODO均为中链甘油酯,具有生物相容性、载水量高、对人体无毒等优点[5]。由于IPM载水量较大,乳化剂需求量相对较小,形成的ME面积大,故最终选择其为油相。大豆磷脂是一种天然的两性离子表面活性剂,具有乳化能力强、生物相容性好和毒性小等优点,亦有促进药物透皮吸收的作用[6]。无水乙醇作为助乳化剂,可通过降低水相的极性,减少表面张力,弥补单用大豆磷脂时油水界面膜流动性差的不足,无水乙醇与大豆磷脂的组合更有利于ME的形成[7]。该研究通过离体鼠皮的渗透实验,考察了不同制剂及浓度对累积渗透量和渗透速率的影响,结果表明,特立氟胺ME的经皮渗透作用明显高于同浓度普通乳剂及水溶液,该结果与相关报道[8]一致。
表3 4种特立氟胺样品的渗透系数
与0.25%特立氟胺ME比较:*P<0.01;与0.1%特立氟胺普通乳剂比较:#P<0.01;与0.1%特立氟胺水溶液比较:▽P<0.01
猜你喜欢
发明与创新(2022年31期)2022-11-03
中国医药科学(2022年5期)2022-05-05
当代水产(2022年2期)2022-04-26
食品安全导刊(2021年24期)2021-11-28
煤矿爆破(2021年4期)2021-03-11
煤矿爆破(2020年3期)2020-12-08
重庆交通大学学报(自然科学版)(2020年11期)2020-11-25
火炸药学报(2017年3期)2017-06-28
火工品(2017年6期)2017-02-01
妇女生活(2014年4期)2014-09-10
