
耳聋突变基因携带孕妇的配偶基因测序研究
2018-10-23 08:18尚晶晶刘正立石亮程
检验医学与临床 2018年20期
曾 黎,尚晶晶,刘正立,石亮程,柳 钐
(1.湖南省长沙市妇幼保健院遗传科 410007;2.河北科技大学生物科学与工程学院,石家庄 050018;3.北京博奥晶典生物技术有限公司,北京 101111)
听力障碍会严重影响人类的生活,根据世界卫生组织(WHO)的分级标准,当听力损失超过40 dB及以上时,定义为耳聋[1]。根据中国人群流行病学调查分析,目前中国人群耳聋基因携带率为5%[2],而目前60%以上的新生儿耳聋是由遗传因素所导致[3]。遗传性耳聋主要分为综合征型耳聋和非综合征型耳聋,其中非综合征型耳聋约占70%[4]。在分子诊断技术的迅速发展以及大力倡导精准医学的背景下,基因芯片检测目前已成为检测遗传性耳聋的主要方法。流行病学研究表明,中国人群的主要遗传性耳聋致病基因是4个,分别为GJB2、SLC26A4、GJB3和MT-RNR1(线粒体12S rRNA)[2]。通过对婚育人群和孕妇群体进行4个耳聋基因的热点突变筛查,实现一级预防和二级预防,达到遗传性耳聋的早发现、早预防、早治疗[5],这也是目前预防耳聋患儿出生最为经济、有效的方法。尤其是对于高危人群,选择产前诊断技术可有效避免遗传性耳聋患儿的出生,为家庭和社会减轻负担。
1 资料与方法
1.1一般资料 受检对象来自2017年4-12月在长沙市妇幼保健院遗传科就诊孕妇(均为耳聋基因携带者)的配偶78例。耳聋基因携带孕妇是从1 951例孕妇中筛查得到,均已进行9项遗传性耳聋基因检测。
1.2方法
1.2.1采样 抽取受检者外周血1~3 mL,乙二胺四乙酸抗凝。
1.2.2测序位点 GJB2基因全长,SLC26A4基因18个外显子区域,GJB3基因全长,MT-RNR1全长。
1.2.3Sanger测序 当孕妇进行耳聋热点突变基因筛查时,若筛查结果为携带者,对孕妇配偶相应的基因进行测序,Sanger测序试验委托北京博奥医学检验所进行,并出具第三方检测报告。
1.2.4产前检测 若夫妻双方为同一耳聋突变基因携带者,经夫妻双方知情同意签字,在B超定位引导下行羊膜腔穿刺术,抽取羊水8~10 mL用于Sanger测序。
2 结 果
2.1孕妇配偶耳聋基因检测情况 在受检的78例孕妇配偶个体中,共检出12例耳聋基因携带者,见表1。
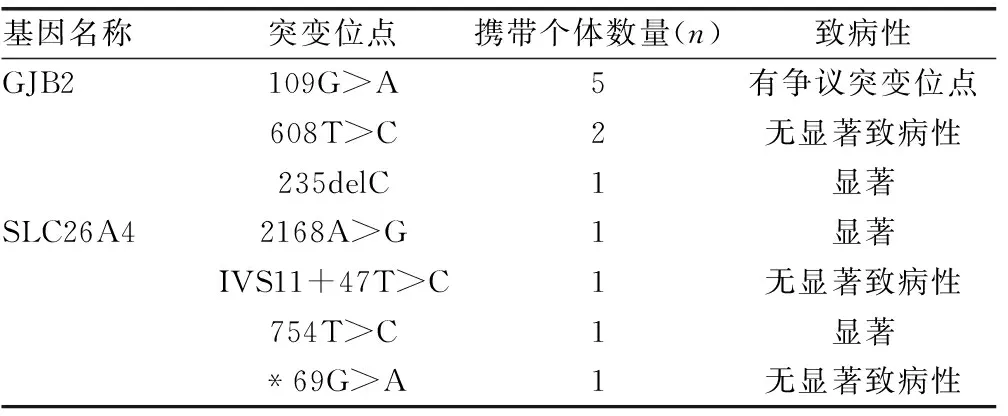
表1 孕妇配偶耳聋基因检测情况
2.2产前诊断 对4对耳聋突变基因携带夫妇进行遗传指导,告知先天性儿聋发生的概率和风险(GJB2和SLC26A4基因突变致聋属于常染色体隐性遗传,生育患儿的风险为25%),4对夫妻均接受了产前诊断,见表2。

表2 4对耳聋突变基因携带夫妇产前诊断情况
注:-表示无数据
3 讨 论
目前临床上对遗传性耳聋缺乏有效的治疗手段,部分耳聋患儿虽借助助听器或人工耳蜗改善听力状况,但大多数因未能及时诊治而影响生活与学习,给整个家庭带来沉重负担[6]。近20年来,我国的出生缺陷率呈上升趋势,目前出生缺陷总发生率约为5.6%,有研究表明,80%的听力障碍患者都是由听力正常的父母所生。
在倡导精准医疗的大背景下,基因芯片技术目前已成为耳聋基因检测的主流技术,性价比也逐渐显现,将基因筛查纳入婚前和孕前检查,实现出生缺陷一级和二级预防[5],是目前临床的主流选择。本研究共对78例耳聋基因携带孕妇的配偶进行相应突变位点测序,发现耳聋基因突变12例,其中109G>A共5例,608T>C共2例,235delC共1例,2168A>G共1例,IVS11+47T>C共1例,754T>C共1例,*69G>A共1例。
109G>A(NM_004004:p.Val 37 Ile)位于GJB2基因EXON2,在109位点G突变为A,氨基酸由缬氨酸(Val)突变为异亮氨酸(Ile),109G>A纯合突变可能与轻中度(25~40 dB)听力损失相关[7],但是目前还具有一定的争议性,未纳入产前诊断。608T>C(NM_004004:p.Ile 203 Thr)位于GJB2基因EXON2,氨基酸由Ile突变为苏氨酸(Thr),该位点比较保守,但是根据已有参考文献与案例,目前认为其属于多态[8]。IVS11+47T>C(NM_000441:c.1341+47T>C)位于SLC26A4基因Intron11,已有文献认为其属于多态[9],但是在耳聋人群中发现比例较高,致病性不明确[10]。754T>C属于显著致病性位点,并且有试剂盒纳入筛查范围[11],本次筛查的754T>C位点也是最先在耳聋先证者中获得。*69G>A位点属于3`UTR区,还未有相关报道,致病性不明确。
GJB2基因在先天性耳聋患者中最为常见,SLC26A4基因在后天突发性耳聋人群中最为常见,与大前庭导水管综合征(LVAS)高度相关。MT-RNR1基因位于线粒体上,临床上主要与药物性耳聋相关,常呈现家族性,一般不作为产前诊断的依据,GJB3基因致病性与临床相关性不是非常明确,目前不作为产前诊断的依据[12-13]。
GJB2基因定位于13q11-12,属于常染色体隐性遗传,DNA全长4 804 bp,编码区长678 bp。GJB2基因编码的缝隙连接蛋白26(Cx26)属于缝隙连接蛋白基因家族,调节K+的运输,Cx26蛋白在人类的耳蜗毛细胞中高表达[14]。突变会导致移码突变,产生无功能蛋白质,使K+回流进入内淋巴液的循环受阻,导致Corti氏器的钾中毒,从而引起感音神经性聋[15]。在本研究中在耳聋基因携带孕妇的配偶中发现两例致病性耳聋突变,1例为父方299delAT杂合,母方235delC杂合,经家属同意,Sanger测序诊断腹中胎儿在测序位点范围内为235delC杂合;1例为父方235delC杂合,母方235delC杂合,经家属同意,Sanger测序诊断腹中胎儿在测序位点范围内为235delC杂合突变。在测序研究过程中有许多受检者还发现109G>A突变,由于临床意义不明确[15],不进行产前诊断。另外还发现79G>A,341A>G多态,在人群中检出率较高,不纳入产前诊断。
SLC26A4基因定位于7q22-31.1,属于常染色体隐性遗传,含有21个外显子,编码含有780个氨基酸的蛋白质Pendrin。Pendrin是一种跨膜蛋白,属于离子转运体26A家族,主要在甲状腺、肾脏和内耳中高表达。研究表明Pendrin主要与Cl-、I-、HCO3-和蔗糖转运有关[16]。此次发现有1例孕妇生育第一胎为听力障碍患者,经过Sanger测序确诊是SLC26A4基因387delC/754T>C复合杂合突变(父方754T>C杂合突变,母方387delC杂合突变)导致听力障碍,经过家属同意,进行羊水穿刺诊断,经过测序诊断腹中胎儿为387delC单杂合突变,预测胎儿听力正常。另外一对夫妇是父方为2168A>G杂合,母方为IVS7-2 A>G杂合,经过测序诊断腹中胎儿在测序位点范围内为野生型,预测听力正常。
对于耳聋发生的高危人群,应明确耳聋病因,实现出生缺陷的一级预防[13],从而减少耳聋出生缺陷,提高人口素质,这对我国妇幼保健工作具有重要的临床意义和研究价值。
猜你喜欢
中国现代医生(2022年19期)2022-11-04
昆明医科大学学报(2022年4期)2022-05-23
中国听力语言康复科学杂志(2021年6期)2021-12-21
中医眼耳鼻喉杂志(2021年1期)2021-07-22
种子(2021年3期)2021-04-12
中国生殖健康(2020年4期)2021-01-18
中国生育健康杂志(2018年6期)2018-11-13
中国生殖健康(2018年4期)2018-11-06
科技视界(2016年27期)2017-03-14
中学生理科应试(2016年7期)2016-05-14
