
基于表面静电排斥/反相混合模式色谱的黄连生物碱分析方法
2018-10-11 05:25张华蓉郭志谋闫竞宇金高娃王联芝
色谱 2018年10期
张华蓉, 郭志谋, 于 伟, 闫竞宇, 金高娃*, 王联芝*
(1. 湖北民族学院, 湖北 恩施 445000; 2. 中国科学院大连化学物理研究所, 辽宁 大连 116023; 3. 中科院大化所中国医药城生物医药创新研究院, 江苏 泰州 225300)
黄连是一种多年生草本类植物,为临床常用药材,基源为毛茛科植物黄连CoptischinensisFranch.、三角叶黄连CoptisdeltoideaC. Y. Cheng et Hsiao或云连CoptisteetaWall.的干燥根茎,分别习称“味连”“雅连”“云连”。现代研究发现黄连的药物活性成分主要是异喹啉类生物碱,目前从黄连中发现的生物碱有30多个[1-6],其中小檗碱(berberine)、黄连碱(coptisine)、巴马汀(palmatine)和药根碱(jatrorrhizine)等占黄连总生物碱的90%左右,小檗碱(又称黄连素)在黄连生物碱中含量最高,占总碱的50%以上[7,8]。研究发现,黄连生物碱可在降血脂、降血糖,治疗糖尿病、肠炎、湿热痞满、泻痢、目赤、牙痛、消渴、痈肿疔疮、湿疹和耳道流脓等方面发挥重要的作用[9-13]。黄连生物碱分离分析方法的建立对于黄连药材的质量标准研究和新药研发具有重要意义。然而,黄连生物碱在常规的反相模式色谱柱上存在色谱峰易拖尾、分离选择性差、载样量低等问题,目前解决这些问题的手段主要有使用高纯度的B型硅胶基质、提高硅胶色谱柱封尾效果[14]、在流动相中添加离子对试剂或缓冲盐[15,16]等。
2015版《中国药典》[1]利用离子对试剂十二烷基硫酸钠作为流动相添加剂(每100 mL中加入0.4 g十二烷基硫酸钠)分析黄连样品,目标生物碱的分离选择性以及色谱峰形都较好。但是,离子对试剂的使用存在以下弊端:色谱柱平衡困难、分析重复性差、对色谱柱损伤严重、与质谱不兼容、配制过程繁杂、价格昂贵等。因此,发展不添加离子对试剂的黄连生物碱分离分析方法十分必要。
为了实现碱性化合物优异的峰形以及选择性,郭志谋等[17]开发了极性共聚技术,制备获得了极性反相色谱柱C18HCE,其代表性表面化学结构见图1。在低pH和低离子强度条件下对碱性化合物的分离有独特的优势[18,19]。研究发现,C18HCE色谱柱对碱性化合物的分离属于表面静电排斥/反相混合模式色谱机理[20],分析对象的保留和选择性不仅受有机相含量的影响,而且受流动相添加剂种类、浓度、pH值等多元化因素影响。随后,Waters公司也推出带有正电荷的CSHC18色谱柱,在低离子强度酸性流动相条件下,对碱性化合物的分离分析同样有效[21,22]。本文以黄连生物碱为研究对象,使用表面正电荷反相色谱柱,通过优化流动相中添加剂的种类及在流动相中的体积分数,实现黄连主要生物碱的对称峰形及良好的选择性,并结合质谱对其主要色谱峰进行识别,最后,利用优化的高效液相色谱分析方法对湖北、重庆两个产地不同批次的黄连药材进行主要生物碱类物质的定量分析。
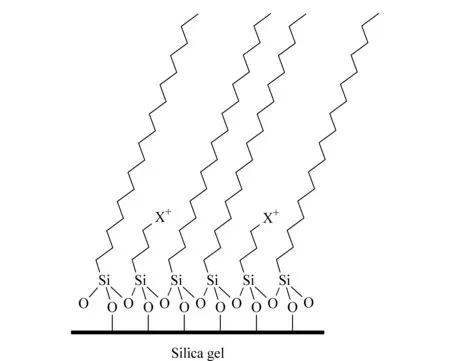
图 1 C18HCE柱的代表性表面化学结构图Fig. 1 Representative surface chemistry structure of the C18HCE material
1 实验部分
1.1 仪器、试剂与材料
Waters Acquity Arc高效液相色谱分析仪(美国Waters公司),配有2998二极管阵列检测器、自动进样器、四元溶剂管理器和Empower工作站;超声波清洗机(宁波新芝生物科技股份有限公司,中国); Waters 2695-QDA液相色谱-质谱联用仪(美国Waters公司)。
甲酸(色谱级,阿拉丁试剂有限公司)、乙酸(分析纯,国药集团化学试剂有限公司)、乙腈(液相色谱纯,上海星可高纯溶剂有限公司)、甲醇(液相色谱纯,德国默克公司)、盐酸(分析纯,国药集团化学试剂有限公司)、娃哈哈纯净水(南京娃哈哈饮料有限公司)、盐酸小檗碱和盐酸药根碱标准品购于中国食品药品检定研究院。
黄连药材:共收集黄连药材6批,分别为湖北统货、湖北大条、湖北单支、重庆统货、重庆大条、重庆单支黄连。湖北黄连购于恩施州利川市汪营黄连市场,四川黄连购于重庆石柱县黄水黄连市场,经中国科学院大连化学物理研究所执业药师杨小平鉴定为黄连(CoptischinensisFranch.)的干燥根茎。
1.2 色谱条件
色谱柱:C18HCE柱(250 mm×4. 6 mm, 5 μm,实验室自制);柱温35 ℃;流动相A为0.1%(体积分数,下同)乙酸水溶液,B为0.1%乙酸乙腈溶液;梯度洗脱条件:0~16 min, 10%B~11%B; 16~30 min, 11%B~60%B; 30~32 min, 60%B~90%B; 32~35 min,保持90%B不变;流速1.0 mL/min;进样体积5 μL;检测波长345 nm。
1.3 质谱条件
电喷雾离子源,正离子模式,质量扫描范围50~1 250 Da,毛细管电压0.8 kV,锥孔电压15 V,离子源温度600 ℃。
1.4 标准溶液的配制
精密称取盐酸小檗碱10 mg,用甲醇溶解得质量浓度为100 mg/L 的盐酸小檗碱对照品溶液。用甲醇稀释对照品溶液,配制成质量浓度为0.5、5、10、40、80 mg/L 盐酸小檗碱对照品溶液。分别取6个质量浓度的对照品溶液,过0.22 μm微孔滤膜于进样瓶中备用。
1.5 供试样品溶液的配制
参考《中国药典》[1]提取方法,取黄连粉末(过20目筛网)约0.2 g,精密称定,置具塞锥形瓶中,加入甲醇-盐酸(100∶1, v/v)混合溶液50 mL,密塞,称定重量,超声处理(功率500 W,频率40 kHz)30 min,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,精密量取续滤液1 mL,置于10 mL量瓶中,加甲醇至刻度,摇匀,过0.22 μm微孔滤膜,取续滤液,即得。
2 结果与讨论
2.1 常规C18柱和C18HCE柱的分析结果对比
在分析碱性化合物时,常以甲酸作为流动相添加剂[23,24],但在甲酸这种简单的流动相添加剂条件下极易出现碱性化合物峰拖尾的现象。本实验分别考察了常规的Welch Ultimate®XB-C18柱(250 mm×4. 6 mm, 5 μm,月旭科技(上海)股份有限公司)和实验室自制的表面正电荷色谱柱C18HCE在甲酸作为流动相添加剂时在各自优化的梯度条件下对同一黄连样品的分离效果,结果见图2。通过与文献[25]中黄连生物碱的紫外光谱图对比,发现常规C18柱所得色谱图的前3个对称峰并非黄连生物碱,后面6个峰为黄连生物碱类物质,但存在明显的拖尾现象;C18HCE柱得到的5个色谱峰为黄连生物碱类物质,虽然选择性差,但峰形对称。这说明即使是使用高纯B型硅胶、封尾效果较好的常规C18柱,在不添加离子对试剂的条件下所得黄连生物碱的峰形依然较差,而C18HCE柱却能在甲酸作为添加剂的条件下明显改善黄连生物碱主要色谱峰的峰形。由于黄连生物碱的保留行为主要受色谱柱固定相表面正电荷与生物碱之间的静电排斥作用的影响,为了优化C18HCE柱对生物碱的选择性,下文将通过优化流动相的添加剂种类及体积分数来调节表面静电排斥作用,以得到主要色谱峰的对称峰形及良好的选择性。
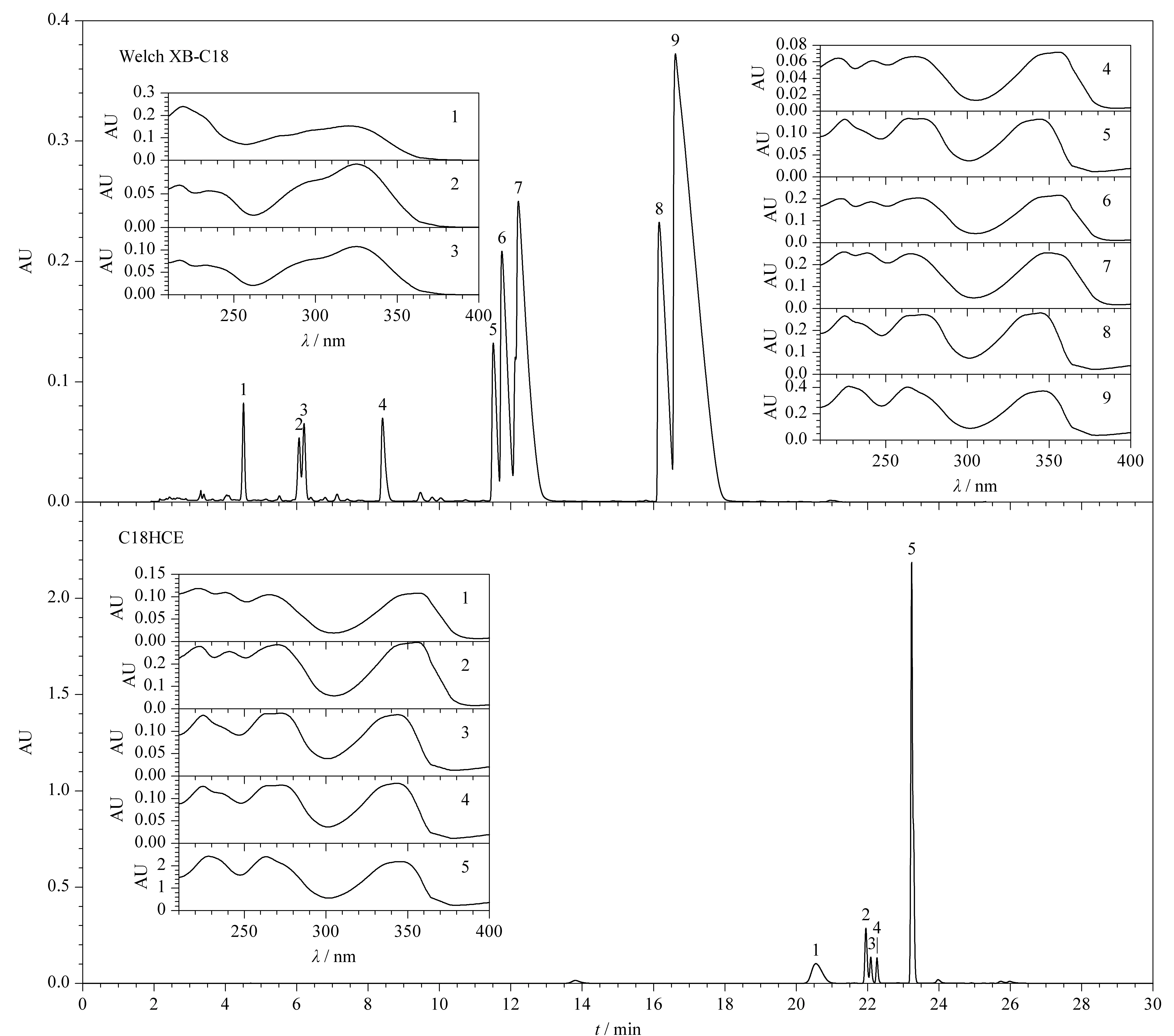
图 2 Welch Ultimate® XB-C18和C18HCE两种色谱柱分析结果对比图Fig. 2 Comparative analysis of Welch Ultimate®XB-C18 and C18HCE column Mobile phase A: water containing 0. 1%(v/v) formic acid; mobile phase B: acetonitrile containing 0. 1%(v/v) formic acid. Gradient program for Welch Ultimate® XB-C18: 0-30 min, 20%B-35%B; 30-32 min, 35%B-90%B; 32-35 min, 90%B. Gradient program for C18HCE: 0-16 min, 10%B-11%B; 16-30 min, 11%B-60%B; 30-32 min, 60%B-90%B; 32-35 min, 90%B. Injection volume: 5 μL. Detection wavelength: 345 nm. Column temperature: 35 ℃. Flow rate: 1.0 mL/min.
2.2 流动相添加剂的优化
由于C18HCE色谱柱适合在低离子强度酸性条件下使用,兼顾与质谱的兼容性,选择色谱分析中常用的甲酸、乙酸作为流动相添加剂,并优化其体积分数,考察两种酸对黄连生物碱的峰形、分离选择性及保留等的影响。
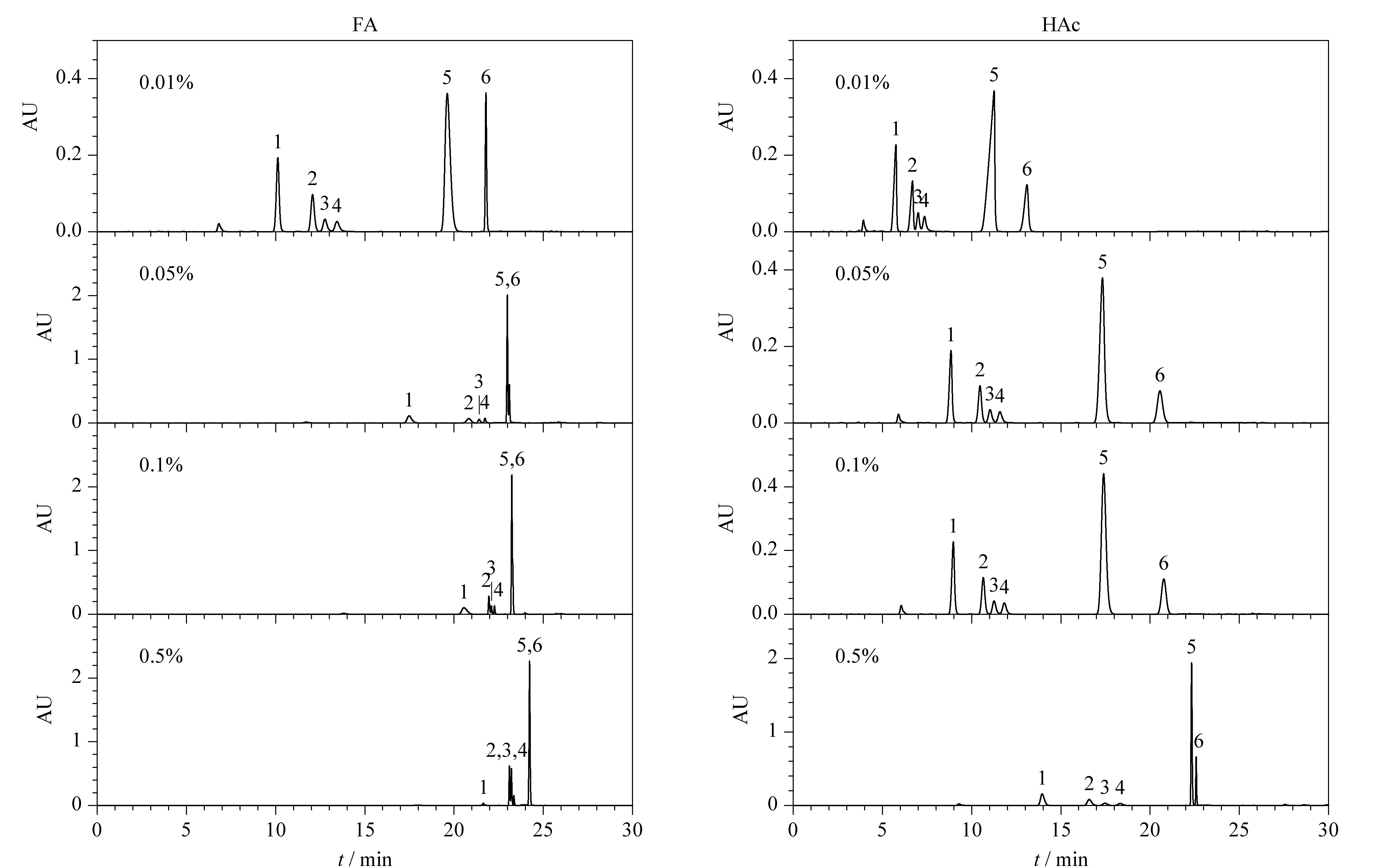
图 3 以不同体积分数的酸作为添加剂时黄连生物碱的色谱图Fig. 3 Chromatograms of Coptis chinensis alkaloids with different volume percentages of acids Mobile phase: A. water; B. acetonitrile. Gradient program: 0-16 min, 10%B-11%B; 16-30 min, 11%B-60%B; 30-32 min, 60%B-90%B; 32-35 min, 90%B. Peaks: 1. coptisine; 2. epiberberine; 3. columbamine; 4. jatrorrhizine; 5. berberine; 6. palmatine.
分别以体积分数为0.01% 、0.05% 、0.1% 、0.5%的甲酸和乙酸为流动相添加剂分析黄连样品,结果如图3所示,可以看出,在这两种酸性添加剂条件下都能得到比较对称的色谱峰形,表明表面正电荷色谱柱C18HCE在酸性添加剂条件下能够实现黄连生物碱主要色谱峰的对称峰形。随着流动相中酸的体积分数增大,目标物的保留增强,这是由于酸的体积分数越大,流动相的离子强度越大,色谱固定相表面正电荷与生物碱之间的静电排斥作用减弱,因此保留增强。整体来看,甲酸作为流动相添加剂时生物碱的保留比乙酸强,这是因为甲酸在溶液中的电离程度比乙酸大,相同体积分数时离子强度更大,所以保留更强。在甲酸条件下,仅0.01%甲酸作为流动相添加剂时分离效果较好,其他含量条件下主要色谱峰之间的选择性都较差,体积分数在0.05%及以上时出现5、6号峰重叠现象,在0.1%及以上时前4个峰也出现了重叠现象。在乙酸作为流动相添加剂时可以得到6个色谱峰,比较各峰之间的分离度、对称性及峰分布可以看出,在0.01%乙酸为添加剂时,峰稍有前伸现象,可能是离子强度太低,静电排斥作用较强导致峰形前伸。在0.5%乙酸为添加剂时,5、6号峰的分离度变差。0.05% 、0.1%乙酸为添加剂时主要色谱峰的峰形及选择性都较好,因此选择0.1%乙酸为流动相添加剂继续后面的实验。
2.3 黄连中主要生物碱类化合物的识别
据文献[6,26]报道,黄连中的生物碱主要为黄连碱、小檗碱、表小檗碱、非洲防己碱、药根碱和巴马汀。在0.1%乙酸作为流动相添加剂的条件下,利用液相色谱-质谱联用进行分析,参考文献[26],结合盐酸药根碱和盐酸小檗碱对照品对所得6个主要色谱峰进行识别,结果见图4。具体识别结果如下:m/z为320.10的1号峰为黄连碱;m/z为336.13的2号峰和m/z为336.14的5号峰是一对同分异构体,通过与盐酸小檗碱标准品的保留时间对比可知5号峰为小檗碱,则2号峰为其异构体表小檗碱; 3号峰和4号峰具有相同的质荷比338.16,是同分异构体,通过与盐酸药根碱标准品的保留时间对比可知4号峰为药根碱,则3号峰为其异构体非洲防己碱;根据m/z为352.20并与文献[26]进行比对,确认6号峰为巴马汀。
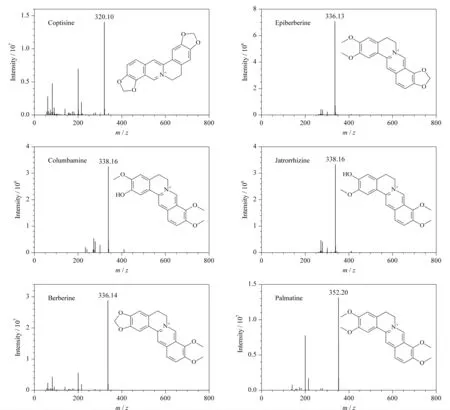
图 4 黄连中主要生物碱的质谱定性分析结果Fig. 4 Qualitative analyses results of main alkaloids in Coptis chinensis by mass spectrometry
2015版《中国药典》[1]中以十八烷基硅烷键合硅胶为填充剂、以乙腈-0.05 mol/L 磷酸二氢钾溶液(50∶50, v/v)(每100 mL中加十二烷基硫酸钠0.4 g,再以磷酸调节pH值为4.0)为流动相实现了黄连药材中黄连碱、表小檗碱、小檗碱、巴马汀4种生物碱的分离与含量测定,本文所建立的方法在此基础上还实现了非洲防己碱、药根碱两种黄连生物碱的分离和识别,对黄连药材质量标准建立、新药研究以及产品开发具有积极作用。同时我们也注意到,对于表面正电荷色谱柱,流动相添加剂微弱的体积分数变化就会对目标物的保留及选择性造成极大的影响,这一点在之前的研究中未见报道。因此,研究这类色谱柱表面相互排斥作用的调节很有价值。
2.4 方法学考察
2.4.1标准曲线、检出限及定量限
参考2015版《中国药典》[1]中对黄连生物碱的含量测定方法,以盐酸小檗碱进行方法学考察。按1.2节色谱条件分析1.4节中配制的不同浓度的盐酸小檗碱对照品溶液,每个浓度样品重复分析3次,以盐酸小檗碱的质量浓度(X, mg/L)为横坐标、峰面积(Y)为纵坐标建立标准曲线。结果表明,小檗碱在0.5~100 mg/L 范围内线性关系良好,线性相关系数(r2)=0.999 6,线性回归方程为Y=6 966.1X+7 922。
以信噪比为3确定检出限为0.17 mg/L,以信噪比为10确定定量限为0.5 mg/L,比文献[27,28]的检出限和定量限更低。
2.4.2精密度试验
取1.4节100 mg/L 的盐酸小檗碱对照品溶液适量,按1.2节色谱条件连续进样测定6次,记录峰面积。结果表明,峰面积的相对标准偏差(RSD)为0.96%(n=6),表明仪器精密度良好。
取湖北大条黄连,按1.5节步骤制备供试样品溶液,按1.2节色谱条件连续进样测定6次,记录峰面积。结果表明,黄连碱、表小檗碱、非洲防己碱、药根碱、小檗碱、巴马汀峰面积的RSD分别为0.93% 、0.66% 、0.95% 、0.93% 、1.09% 、1.12%(n=6),说明方法精密度良好。
2.4.3重复性实验
称取5份湖北大条黄连,按1.5节方法制备供试样品溶液,按1.2节色谱条件测定,记录峰面积。结果表明,黄连碱、表小檗碱、非洲防己碱、药根碱、小檗碱、巴马汀峰面积的RSD分别为1.29% 、1.55% 、2.61% 、4.20% 、2.14% 、1.49%(n=5),说明方法的重复性良好。
2.4.4稳定性实验
取1.5节供试品溶液适量,分别于室温下放置0、4、8、24、36 h,按1.2节色谱条件进样测定。结果表明,黄连碱、表小檗碱、非洲防己碱、药根碱、小檗碱、巴马汀峰面积的RSD分别为1.06% 、0.60% 、1.22% 、1.12% 、0.86% 、0.92%(n=5),表明供试品溶液室温放置36 h稳定性良好。
2.4.5回收率试验
按1.5节处理黄连样品6份,处理前分别加入100 mg/L 盐酸小檗碱对照液0.31 mL,按1.2节色谱条件进样测定,小檗碱峰面积的RSD为3.40%(n=6),平均加标回收率为93.74% 。
2.5 样品测定
湖北和重庆两地是黄连药材中味连的主产地,产量大、质量好。按1.5节方法分别制备湖北统货黄连、湖北大条黄连、湖北单支黄连、重庆统货黄连、重庆大条黄连、重庆单支黄连样品,每批药材平行制备3份,在1.2节条件下进行分析。参照2015版《中国药典》[1]中关于黄连中物质含量测定的方法,以盐酸小檗碱对照品的峰面积为对照计算各生物碱的含量。结果如表1所示:不同批次的黄连药材中生物碱的含量有所差别,黄连总生物碱占黄连成分的10.64% ~16.03% ,其中小檗碱的含量约占总碱的56.89% ~58.86% 。湖北大条黄连中总碱含量占药材的16.03% ,其中小檗碱含量占药材的9.12% ,在这6批药材中最高。总碱含量从高到低依次为湖北大条黄连、湖北单支黄连、重庆单支黄连、湖北统货黄连、重庆大条黄连、重庆统货黄连,小檗碱含量从高到低依次为湖北大条黄连、重庆单支黄连、湖北单支黄连、湖北统货黄连、重庆大条黄连、重庆统货黄连。文献[29,30]中研究了不同产地黄连的指纹图谱及质量研究,所得结果表明不同产地的黄连总生物碱的含量存在差异,同时也显示出湖北利川黄连中总生物碱的含量比重庆石柱的高。我们所得结果与其一致。
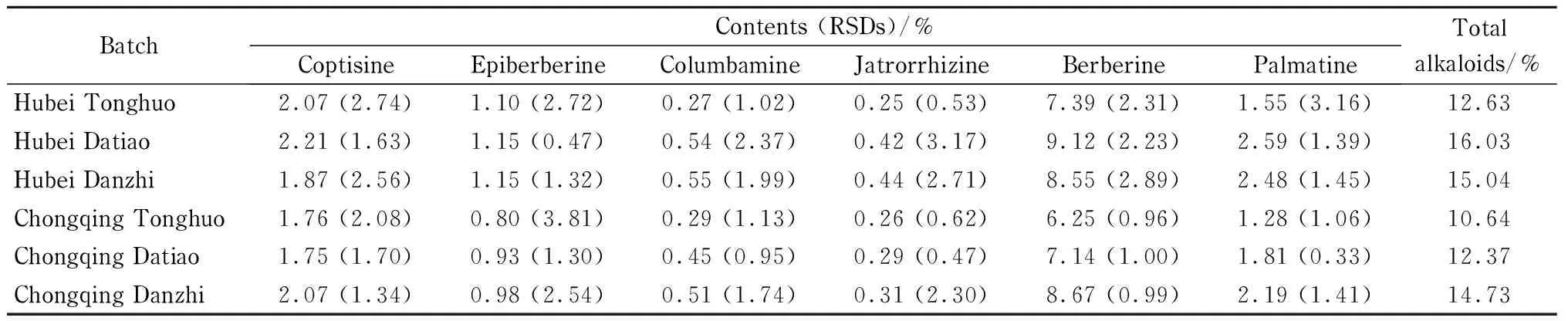
表 1 不同批次黄连药材中主要生物碱的含量及RSD(n=3)
3 结论
本文以黄连药材为研究对象,利用表面正电荷反相色谱柱C18HCE,通过优化流动相中添加剂的种类及体积分数,确定了以0.1%乙酸作为流动相添加剂的基于表面静电排斥/反相混合模式色谱的黄连生物碱分析方法,并结合质谱检测器识别了黄连中6种主要生物碱,分别为黄连碱、表小檗碱、非洲防己碱、药根碱、小檗碱、巴马汀。该方法可以实现黄连生物碱色谱峰的良好分离及对称峰形,稳定性好,重复性好,能够有效地应用于实际黄连药材中生物碱的分离分析与含量测定,为碱性化合物的分离和定量分析提供了新的思路和方法。此外,不同于普通的C18柱,表面正电荷色谱柱对目标物的保留行为受流动相中有机相比例、添加剂种类及体积分数等多种因素影响,流动相中离子强度的微弱变化都可能引起色谱柱表面电荷状态的极大变化,从而对保留行为产生很大影响,因此该色谱柱的相关性能还需进一步考察。
猜你喜欢
现代临床医学(2021年4期)2021-07-31
西南农业学报(2020年8期)2020-12-10
工程质量(2019年8期)2019-11-15
药学研究(2019年5期)2019-06-05
世界科学技术-中医药现代化(2018年9期)2019-01-29
中成药(2018年12期)2018-12-29
中成药(2018年3期)2018-05-07
中成药(2017年12期)2018-01-19
中成药(2017年7期)2017-11-22
中成药(2017年8期)2017-11-22
