
大孔硅胶的制备及其在多糖衍生物手性固定相上的应用
2018-10-11 07:45吴海波薛兴亚李奎永周永正
色谱 2018年10期
吴海波, 薛兴亚, 李奎永, 周永正*
(1. 浙江华谱新创科技有限公司, 浙江 温岭 317503; 2. 中国科学院大连化学物理研究所, 中国科学院分离分析化学重点实验室, 辽宁 大连 116023)
近年来,随着高效液相色谱技术的发展,特别是工业制备色谱的推广与应用,硅胶基质填料的液相制备技术越来越成熟[1-3]。孔径8~10 nm的硅胶基质在色谱上应用最为普遍,制备工艺也相对简单、稳定。但在分离蛋白质、多肽、聚合物等大分子物质时,为了让样品可以进入孔内与键合相接触,往往需要采用由更大孔径的硅球制备的固定相[4,5]。另外,一些由聚合物涂覆或键合制备的固定相也需要使用大孔径的硅胶做基质[6,7]。
在手性分离领域,多糖衍生物型固定相是应用最广的一类手性固定相,可解决90%以上的手性分离问题。此类固定相由直链淀粉或纤维素的衍生物涂覆或键合到硅胶表面制得。多糖衍生物的聚合度大约200~300,所用的基质通常为孔径100 nm的大孔硅胶[7]。目前,手性制备分离成本较高,主要因为填料较为昂贵。在填料成本中,大孔硅胶基质也占据相当部分的比例。
大孔硅胶可由小胶粒直接烧结制得,但机械稳定性一般较差或孔径单一,不易控制。目前,大孔硅胶多以廉价的小孔径硅胶作为原料,采用一定的扩孔方法进行制备。扩孔方法一般有两种:热液法和焙烧法[8]。热液法是将硅胶分散于水或氨水溶液中,于高压釜内进行高温扩孔处理;焙烧法是先将硅胶浸渍在NaCl、LiCl或KNO3等无机盐配制的复盐溶液中,取出烘干后,混合物经高温焙烧进行扩孔[9-11]。
本研究考察并对比了两种扩孔方式的扩孔效果,将所制得的100 nm大孔硅胶分别涂覆纤维素-三(3,5-二甲基苯基氨基甲酸酯)制成手性固定相,并评价了手性分离能力。
1 实验部分
1.1 仪器、试剂与材料
实验所用球形硅胶,平均粒径5 μm,孔径10 nm,由浙江华谱新创科技有限公司提供。
微晶纤维素为化学纯,购自国药集团化学试剂有限公司。3,5-二甲基苯基异氰酸酯、3-氨丙基三甲氧基硅烷、NaCl、LiCl·H2O、四氢呋喃、吡啶、无水乙醇等试剂为分析纯,购自阿拉丁试剂(上海)有限公司。
分析用高效液相色谱为Waters 515泵系统,配7725i手动进样器、2489检测器(美国Waters公司)。JXR1200-30a马弗炉(上海均珂仪器公司)。Tristar Ⅱ 3020比表面分析仪(美国Micromeritics Instrument公司)。Elementar Vario EL Ⅲ元素分析仪(德国Elementar Analysensysteme公司)。
1.2 实验方法
1.2.1热液扩孔
将160 g硅胶分散于300 mL 22 g/L 氟化钠水溶液中,搅拌均匀,倒入500 mL高压釜中,密封后置于烘箱内,加热至设定温度保持一段时间。自然冷却,过滤,纯水洗涤至中性,50 ℃真空干燥。
1.2.2焙烧扩孔
将11.25 g LiCl·H2O和7.5 g NaCl溶于200 mL水中,超声溶解后加入100 g硅胶,超声10 min,置于150 ℃烘箱中烘干。之后,置于马弗炉中焙烧5 h,取出。冷却至室温,加水分散,过滤,水洗,50 ℃真空干燥。
1.2.3硅胶氨基化
将50 g扩孔后的硅胶分散于250 mL 2.5 mol/L HCl溶液中于100 ℃回流6 h,过滤,水洗至中性,干燥。硅胶分散于200 mL甲苯中,加入10 mL 3-氨丙基三甲氧基硅烷,于110 ℃回流反应16 h,过滤,用甲醇洗涤,于50 ℃真空干燥。元素分析结果:热液扩孔,N 0.20% , C 0.87% , H 0.36% ;焙烧扩孔,N 0.18% , C 0.81% , H 0.33% 。
1.2.4纤维素衍生物的合成与涂覆
将5.0 g微晶纤维素分散于100 mL无水吡啶中,加入18.1 g(4倍于纤维素中羟基的物质的量)3,5-二甲基苯基异氰酸酯,于100 ℃下反应24 h,向反应体系中加入甲醇,析出沉淀,过滤,于50 ℃真空干燥得15.0 g固体。元素分析结果:N 6.94% , C 65.36% , H 5.88% 。凝胶渗透色谱(GPC)测得重均相对分子质量Mw=152 123,聚合度约250。
取4.0 g纤维素-三(3,5-二甲基苯基氨基甲酸酯)溶解于100 mL四氢呋喃中,加入16.0 g氨基化的大孔硅胶,拌匀,于旋蒸仪上50 ℃缓慢旋干(约3 h)。在旋干后的固定相中加入120 mL乙醇,超声分散,80 ℃搅拌回流3 h,过滤,乙醇洗涤,于50 ℃真空干燥。元素分析:热液扩孔,N 1.25% , C 11.18% , H 1.43%(实际涂覆量18.0% ,涂覆率90% );焙烧扩孔,N 1.34% , C 12.43% , H 1.66%(实际涂覆量19.2% ,涂覆率96% )。
1.2.5色谱柱的装填与评价
固定相加甲醇匀浆,于50 MPa压力下进行装柱(250 mm×4.6 mm),甲醇顶替15 min。
系统死时间t0由1,3,5-三叔丁基苯进行测定。保留因子k=(t-t0)/t0;分离因子α=k2/k1;分离度Rs=1.18×[(t2-t1)/(W1+W2)],其中W1与W2表示两峰的半峰宽。
手性分离条件:流速,1.0 mL/min;温度,25 ℃;检测波长,220 nm或254 nm。流动相:分析物1~6,正己烷-异丙醇(90∶10, v/v);分析物7~9,正己烷-异丙醇(80∶20, v/v);分析物10,正己烷-异丙醇-三氟乙酸-二乙胺(90∶10∶0.2∶0.1, v/v/v/v);分析物11,正己烷-异丙醇-三氟乙酸-二乙胺(80∶20∶0.2∶0.1, v/v/v/v)。
稳定性试验考察:样品,分析物1;流动相,100%甲醇;温度,30 ℃;流速,0.8 mL/min;检测波长220 nm;进样次数,100针。
2 结果与讨论
2.1 热液法扩孔
热液法扩孔是基于硅胶骨架的Si-O-Si键在高温、高压的水蒸气中不断水解和再缩聚,从而达到扩孔效果[8]。通常,扩孔时会在水中加入少量氨水提供碱性条件,以促进细小基本粒子的溶解和在大基本粒子上的缩聚。由于氨水易挥发,添加量不易控制。本实验中,在水中加入了少量氟化钠来提供弱碱性环境,另外水解生成的少量氢氟酸也可以增强对细小基本粒子的溶解性。
表1列举了3个条件下水热扩孔的结果,图1给出了其中2号条件下扩孔后的硅胶与Fuji-1000硅胶的扫描电镜图。可以看出,纯水条件下扩孔较慢。160 ℃加热48 h后,比表面与孔径变化均很小。相同条件下,22 g/L 氟化钠中硅胶扩孔则接近100 nm。扩孔40 h后,比表面与孔径变化已相对缓慢。由图1可看出,热液扩孔的硅胶孔道不均匀,孔径跨度较大。

表 1 热液法扩孔条件与结果
*Fuji-1000 is commercial 100-nm silica from Fuji Silysia Chemical Ltd.
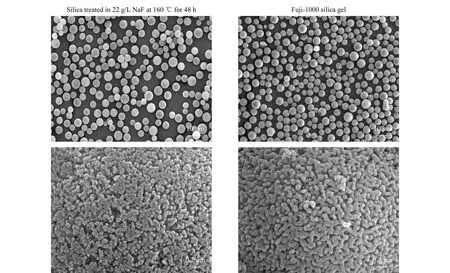
图 1 22 g/L NaF中160 ℃扩孔48 h的硅胶以及Fuji-1000硅胶的扫描电镜(SEM)图Fig. 1 Scanning electron microscopy (SEM) images of silica treated in 22 g/L NaF at 160 ℃ for 48 h and Fuji-1000 silica gel
此外,实验发现热液扩孔后的硅胶夹杂大量硅胶细屑,筛除后硅胶有约10%损失。可能是扩孔过程中一些基质粒子溶解后在溶液中发生了再缩聚。
2.2 焙烧法扩孔
焙烧扩孔是基于高温条件下对原有孔结构的破坏,细小的SiO2质点在复盐的助熔作用下进一步“熔结”长大,从而获得大孔结构[12]。常用的复盐为NaCl、LiCl和KNO3组成的三元复盐体系。通常在10~200 nm范围内,NaCl+LiCl是比较好的复盐体系。本实验选择该体系对焙烧温度、时间和盐量进行了考察。具体焙烧条件和结果见表2。

表 2 焙烧法扩孔条件与结果
*The masses of LiCl·H2O and NaCl indicate amounts added in per 10 g silica.

图 2 焙烧扩孔后的硅胶扫描电镜图Fig. 2 SEM images of silica treated by baking No. 6 in Table 2.
在表2 No. 1和No. 2条件中,每10 g硅胶加入2.25 g LiCl·H2O和1.5 g NaCl,在520 ℃下焙烧。结果表明,扩孔速度较快,1 h便达到了110 nm; 3 h后,平均孔径已至210 nm。由于扩孔较快不易控制,No. 3条件将加入的盐量减半。3 h后,平均孔径达到120 nm,扩孔速度明显降低。之后降低了扩孔温度,No. 4、5、6条件中,将焙烧温度降为500 ℃,可以看出焙烧2、3、5 h的结果比较接近,比表面变化较小,平均孔径为90~100 nm。因此,最佳扩孔条件确定为:每10 g硅胶加入1.125 g LiCl·H2O和0.75 g NaCl, 500 ℃焙烧3~5 h。该条件扩孔结果稳定,便于实际操作控制。
图2为表2中No. 6条件扩孔后硅胶的扫描电镜图。从形态上看,该大孔硅胶与商品化的Fuji-1000硅胶非常相似。孔道较热液法扩孔的硅胶更均匀。
此外,实验发现焙烧法扩孔后的硅胶基本无质量损失,也无硅胶细屑产生。因而,后处理也较热液扩孔法简单。这表明,焙烧扩孔时SiO2质点的“熔结”均在硅胶表面进行,未导致硅胶粒子脱落。
2.3 手性固定相的制备与评价
基于大孔硅胶涂覆纤维素-三(3,5-二甲基苯基氨基甲酸酯)的手性固定相是目前选择性最广的手性固定相之一,由于合成、制备简单,是许多实验研究的对象[13]。本实验分别选取热液法和焙烧法扩孔得到的100 nm硅胶进行氨基化修饰,之后涂覆纤维素-三(3,5-二甲基苯基氨基甲酸酯),制备了相应的手性固定相CSP 1与CSP 2。
热液法扩孔的硅胶涂覆后有明显的颗粒团聚现象,应该是由于硅胶表面小孔较多,一些纤维素衍生物未能进入孔道,在溶液中发生了析出,导致一些填料颗粒相互粘连。乙醇回流处理后,一些仍不分散的小颗粒被弃除。焙烧法扩孔的硅胶涂覆后分散性良好,几乎无团聚颗粒。从元素分析结果中也可以看出,焙烧法扩孔的硅胶涂覆量稍微高一些。由扫描电镜图(图3)可以看出, CSP 2明显具有较好的涂覆均匀性。
CSP 1与CSP 2装柱(250 mm×4.6 mm)后,经检测前者死时间为2.6 min,后者死时间为2.9 min。前者的死体积更小,表明热液法扩孔硅胶的孔隙率偏低。
图4是11种手性化合物的结构,表3是固定相CSP 1与CSP 2对这些对映体的分离结果。
从表3中的结果可以看出,CSP 1与CSP 2对于样品的保留均非常接近,但对于大部分分析物,CSP 2的选择性明显较CSP 1高。例如,化合物7和9在CSP 1上的分离微弱,但在CSP 2上可以得到基线分离;化合物6、10在CSP 2上的分离因子比CSP 1明显大(差值>0.5)。而一些分析物即使在CSP 1与CSP 2上的分离因子相近,后者的分离度也明显高于前者,如化合物2、4的手性分离。化合物7、10、4的分离色谱图见图5。

*CSP 1 based on hydrothermally treated silica; CSP 2 based on baking-treated silica.
CSP 2由于涂覆更均匀,装柱柱效较高,因而对许多对映体的选择性更好,分离度更高。而CSP 1由于固定相涂层不均匀,柱效偏低,分离效果往往较差。另外,CSP 1中由于大分子涂层无法进入小孔,导致其内部裸露的氨基会对样品产生一些非对映选择性的作用[11],从而削弱了固定相对一些对映体的选择性。
为了考察CSP 2的分离稳定性,以化合物1为例,100%甲醇做流动相,连续进样了100针。分离具有很好的重复性(见图6),表明固定相稳定性较好。
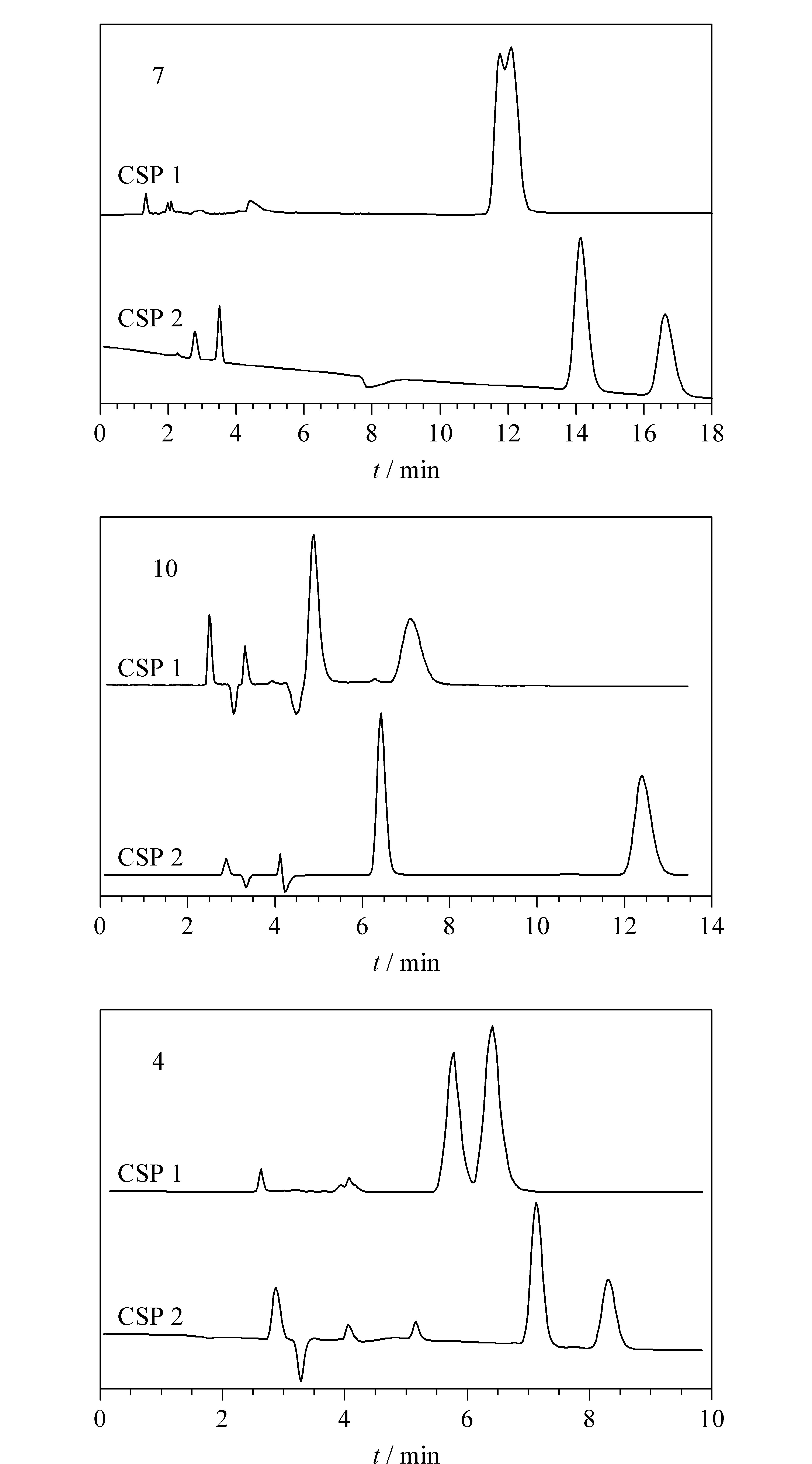
图 5 化合物7、10、4在CSP 1与CSP 2上的分离谱图Fig. 5 Chromatograms of compounds 7, 10 and 4 separated on CSP 1 and CSP 2
总的来看,焙烧扩孔法更为简单、高效。扩孔后的硅胶孔径分布较均匀,比较适合多糖衍生物涂覆。所制得的手性固定相选择性好,装柱柱效高,有利于手性样品的分离与分析。

图 6 化合物1在CSP 2上的重复进样谱图Fig. 6 Chromatograms of repeated injections of compound 1 on CSP 2
3 结论
为制备100 nm大孔硅胶,本文考察了热液法和焙烧法两种扩孔方式。水热法扩孔时,水溶液中加入氟化钠可以增强扩孔效果,但所制得的大孔硅胶孔径不均匀。焙烧法扩孔时,通过调节焙烧温度、时间以及复盐LiCl-NaCl的加入量可以有效控制扩孔效果。该方法简单、高效,扩孔后的硅胶孔径分布更加均匀。硅胶经氨基修饰后涂覆纤维素-三(3,5-二甲基苯基氨基甲酸酯)制得的手性固定相表现了较好的分离选择性与较高的柱效,且稳定性较好,有利于手性样品的分离与分析。
猜你喜欢
分子催化(2022年1期)2022-11-02
天津科技大学学报(2021年1期)2021-02-25
上海计量测试(2020年1期)2020-03-18
雷达学报(2018年1期)2018-04-04
国外医药(抗生素分册)(2016年4期)2016-07-12
国外医药(抗生素分册)(2016年2期)2016-07-12
浙江大学学报(工学版)(2016年2期)2016-06-05
化工进展(2015年3期)2015-11-11
环境科技(2015年1期)2015-11-08
电子世界(2015年24期)2015-01-16
