
基于气相色谱法的帕瑞昔布钠中残留溶剂测定
2018-10-10 03:09:46聂忠莉毛玉萍王晓玲萧茂玲黄金玉
成都大学学报(自然科学版) 2018年3期
聂忠莉, 毛玉萍, 王晓玲, 萧茂玲, 张 勇, 黄金玉, 郭 瑞
(1.成都大学 药学与生物工程学院, 四川 成都 610106; 2.成都克莱蒙医药科技有限公司, 四川 成都 610041)
0 引 言
帕瑞昔布钠,是伐地昔布的水溶性前体,其存在形式为钠盐,主要用于手术后疼痛的短期治疗以及中度或重度术后急性疼痛的治疗.帕瑞昔布钠作为第一代可肌注和静脉注射使用的选择性COX-2抑制剂,于2008年在我国上市.临床试验表明,其对手术术后的恢复效果较佳,且副反应较小,有利于需要经非胃肠给药的手术患者的恢复[1-2].
通过对帕瑞昔布钠的合成工艺以及相关合成文献[3-4]和专利[5]分析发现,其在合成及精制的过程中使用的有机溶剂包括乙醇、正己烷、二氯甲烷、乙酸乙酯、三乙胺、4-二甲氨基吡啶.根据各溶剂性质和沸点的不同,本研究通过参考帕瑞昔布钠的相关标准以及《中国药典》2015年版四部中气相色谱法操作、残留溶剂测定法和药品质量标准分析方法验证指导原则,建立了帕瑞昔布钠残留溶剂的气相色谱测定方法,并对其进行了方法学验证.
1 材料与仪器
1.1 材 料
实验所用材料包括:帕瑞昔布钠(批号,20171201、20171202、20171203),购自成都克莱蒙医药科技有限公司;N,N-二甲基甲酰胺(批号,141171101,HPLC级)、正己烷(批号,121171202,HPLC级),购自上海星可高纯溶剂有限公司;无水乙醇(批号,2015051401,HPLC级)、二氯甲烷(批号,2017091901,HPLC级),购自成都科龙化工试剂厂;乙酸乙酯(批号,20150128,HPLC级)、三乙胺(批号,20170925,HPLC级),购自天津科密欧化学试剂有限公司;4-二甲氨基吡啶(批号,20171113-2629,HPLC级),购自上海易势化工有限公司.
1.2 仪 器
实验所用仪器包括:Agilent 7890A气相色谱仪(美国Agilent公司),BP-210D型电子天平(德国Sartorius公司)等.
2 方法与结果
2.1 溶液的配制
2.1.1 供试品溶液.
取本品约100 mg,精密称定,置顶空瓶中,加入N,N-二甲基甲酰胺1.0 mL,溶解,摇匀,作为供试品溶液.
2.1.2 对照品溶液.
取无水乙醇、正己烷、二氯甲烷、乙酸乙酯、三乙胺、4-二甲氨基吡啶适量,分别加N,N-二甲基甲酰胺溶解并稀释,制成每1 mL约含0.55 mg的乙醇、0.029 mg的正己烷、0.06 mg的二氯甲烷、0.5 mg的乙酸乙酯、0.032 mg的三乙胺、0.5mg的4-二甲氨基吡啶溶液,作为对照品溶液.
2.2 色谱条件的选择
由于三乙胺具有较强的碱性,且4-二甲氨基吡啶沸点(211 ℃)较高,不宜与其他溶剂一起采用气相色谱法进行检测,因此将三乙胺和4-二甲氨基吡啶分别单独进行质量控制研究. 由预实验结果可知(见表1),在色谱条件3下,乙醇、正己烷、二氯甲烷、乙酸乙酯的分离度更好,故选取色谱条件3来检测本品中的乙醇、正己烷、二氯甲烷、乙酸乙酯.另外,在不同柱温下三乙胺的保留时间变化不大,但随温度的升高,理论塔板数不断增加,当柱温为210 ℃其理论塔板数达到100 000以上,拖尾因子为1.016,且空白无干扰,故选取色谱条件6作为本品三乙胺的检查条件.虽然色谱条件7和色谱条件8峰型均较好,但后者的出峰时间比前者提前了将近5 min,且空白均无干扰,故选取色谱条件8作为本品4-二甲氨基吡啶的检查条件.
表1帕瑞昔布钠残留溶剂分析方法色谱条件比较
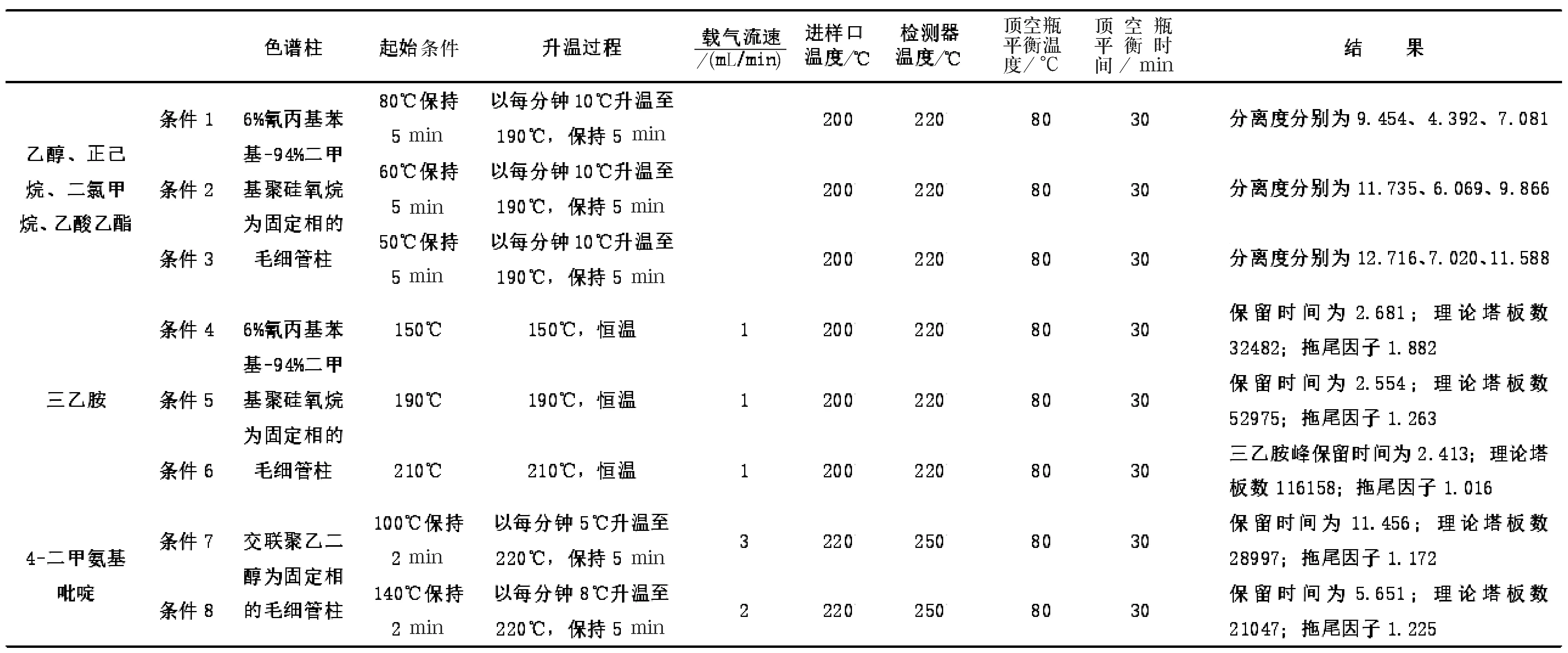
2.3 系统适用性
取无水乙醇、正己烷、二氯甲烷、乙酸乙酯对照品溶液各0.25 mL,置于顶空瓶中,作为乙醇、正己烷、二氯甲烷、乙酸乙酯的混合溶液.在色谱条件3下,分别取对照品乙醇溶液、正己烷溶液、二氯甲烷溶液、乙酸乙酯溶液和混合溶液顶空进样,另取空白溶剂同法进样,记录色谱图(见图1、2).结果显示,乙醇、二氯甲烷、正己烷、乙酸乙酯依次出峰,且色谱峰之间均能完全分离、空白溶剂对测定无干扰.
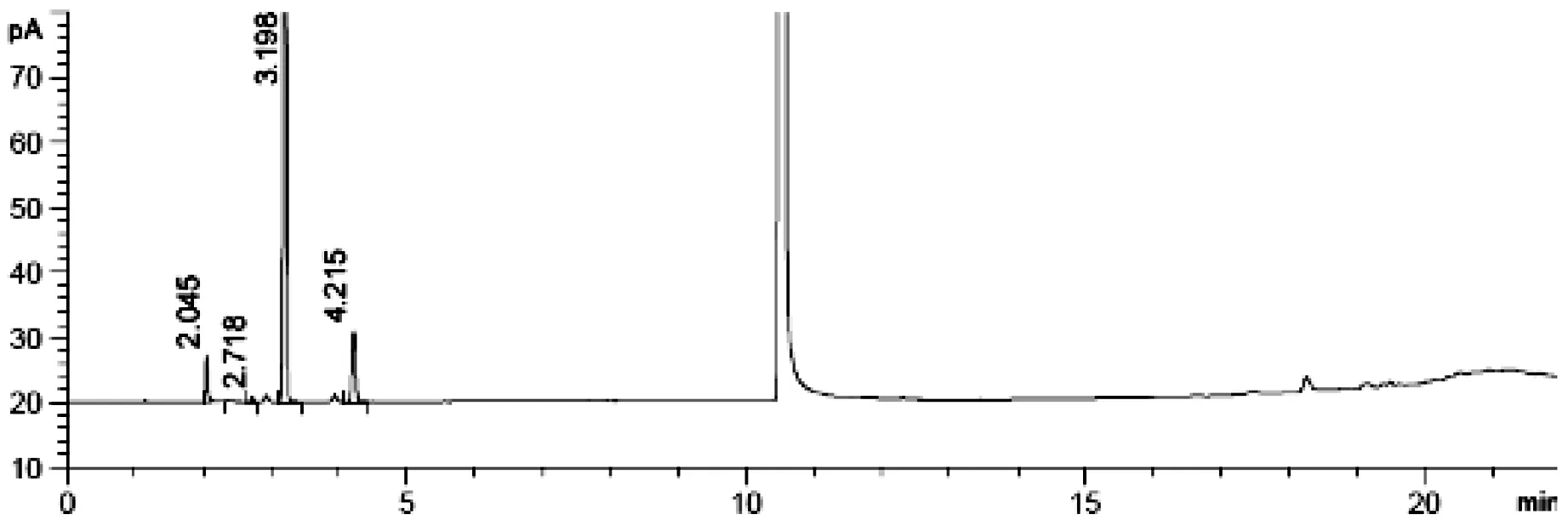
图1乙醇、正己烷、二氯甲烷、乙酸乙酯系统适用性色谱图
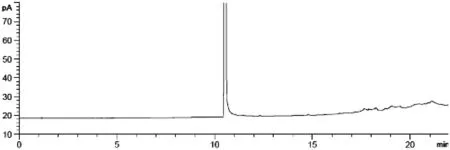
图2 N,N-二甲基甲酰胺空白色谱图
2.4 检测限
取无水乙醇、正己烷、二氯甲烷、乙酸乙酯、三乙胺、4-二甲氨基吡啶各适量,分别加N,N-二甲基甲酰胺溶解并稀释制成一定浓度的溶液,顶空进样,记录色谱图,以信噪比为3时(S/N=3)的浓度作为最低检测限浓度,以信噪比为10时(S/N=10)的浓度作为定量检测限浓度,结果见表2.
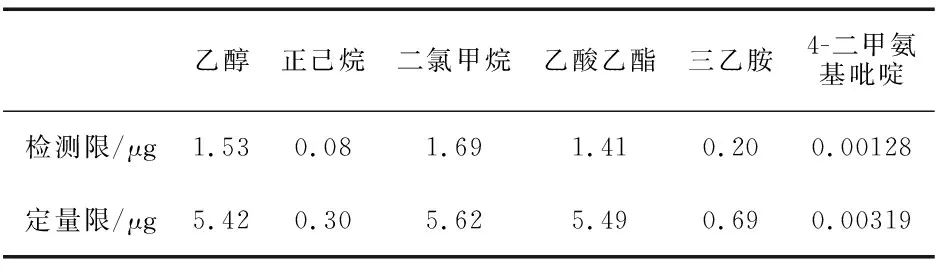
表2 帕瑞昔布钠残留溶剂检测限、定量限实验
2.5 线性与范围
分别精密量取无水乙醇、正己烷、二氯甲烷、乙酸乙酯、三乙胺、4-二甲氨基吡啶适量,加入N,N-二甲基甲酰胺溶解并稀释成适宜的浓度,分别作为线性溶液1、2、3、4、5、6.分别精密量取上述供试品溶液各1 mL,置顶空进样瓶中,密封,顶空进样,记录色谱图,以各溶剂浓度对峰面积进行线性回归并计算相关系数.结果表明:在0.00965~6.372 mg/mL范围内,乙醇(Y=101.27X+2.5014,R2=0.9997)、正己烷(Y=2647.6X+1.896,R2=0.9997)、二氯甲烷(Y=80.325X+0.5648,R2=0.9998)、乙酸乙酯(Y=207.53X+10.242,R2=0.9995)、三乙胺(Y=363.18X+2.4124,R2=0.9994)、4-二甲氨基吡啶(Y=1045.9X-35.25,R2=0.9994)的峰面积与浓度呈良好的线性关系.
2.6 重复性
取帕瑞昔布钠约1.0 g,分别取无水乙醇、正己烷、二氯甲烷、乙酸乙酯、三乙胺、4-二甲氨基吡啶对照品溶液10 mL使其溶解,摇匀后,分别精密量取1 mL 6份,分别置顶空进样瓶中,密封,顶空进样,记录色谱图,计算出无水乙醇、正己烷、二氯甲烷、乙酸乙酯、三乙胺、4-二甲氨基吡啶结果的RSD分别为0.78%、1.97%、1.15%、0.41%、2.27%、1.35%.结果表明,方法的重复性良好.
2.7 稳定性
取帕瑞昔布钠约1.0 g,分别取无水乙醇、正己烷、二氯甲烷、乙酸乙酯、三乙胺、4-二甲氨基吡啶对照品溶液10 mL使其溶解,摇匀后,分别精密量取1 mL 5份,分别置顶空进样瓶中,密封,分别在0、2、4、6、8 h顶空进样,记录色谱图,计算样品中乙醇、正己烷、二氯甲烷、乙酸乙酯、三乙胺、4-二甲氨基吡啶残留量相对标准偏差(见表3).结果表明,溶液在8 h内稳定性良好.

表3 帕瑞昔布钠残留溶剂稳定性实验(n=6)
2.8 回收率
取帕瑞昔布钠(批号,20171201)各约0.1 g(共11份),精密称定,分别置顶空进样瓶中,分别精密加入无水乙醇、正己烷、二氯甲烷、乙酸乙酯、三乙胺、4-二甲氨基吡啶的80%对照品溶液、100%对照品溶液、120%对照品溶液1.0 mL各3份,使样品溶解,分别作为80%、100%、120%的供试品溶液;剩余2份加入1.0 mL空白溶剂使溶解,作为供试样品溶液.分别取对照品溶液、供试品溶液、供试样品溶液顶空进样,记录色谱图,按外标法计算无水乙醇、正己烷、二氯甲烷、乙酸乙酯、三乙胺、4-二甲氨基吡啶测得量及回收率(见表4).结果表明,残留溶剂回收率满足要求.
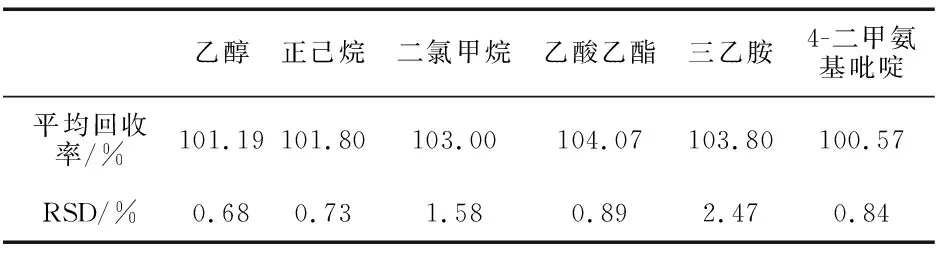
表4 帕瑞昔布钠残留溶剂平均回收率
2.9 耐用性
取帕瑞昔布钠(批号,20171201)约0.1 g,精密称定,置量瓶中,分别加入1.0 mL无水乙醇、正己烷、二氯甲烷、乙酸乙酯、三乙胺、4-二甲氨基吡啶对照品溶液使溶解,摇匀,作为供试样品溶液.分别取上述对照品溶液、供试品溶液1 μL进样,微小改变色谱条件,测定,记录色谱图,按外标法计算无水乙醇、正己烷、二氯甲烷、乙酸乙酯、三乙胺、4-二甲氨基吡啶含量(见表5).结果表明,方法的耐用性良好.
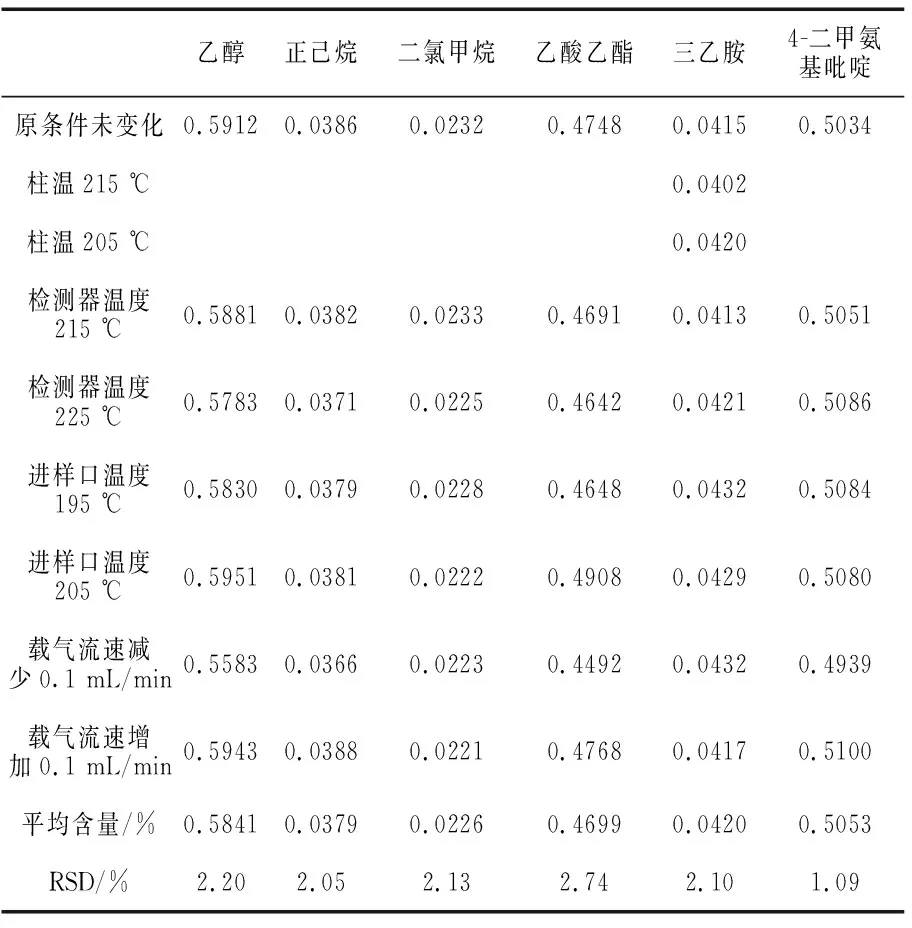
表5 帕瑞昔布钠残留溶剂耐用性实验
2.10 无水乙醇、正己烷、二氯甲烷、乙酸乙酯检测
分别取3批帕瑞昔布钠按“2.1.1"项下方法制得供试品溶液,按“2.1.2"项下方法制得对照品溶液,精密量取供试品溶液与对照品溶液,分别以相应的条件顶空进样,记录色谱图,按外标法计算各溶剂残留量.结果显示,批号20171201和20171202中的乙醇含量为0.16%,批号20171203中的乙醇含量为0.18%,其他有关物质在3批试样中均未检出.
3 讨论与结论
本研究采用气相色谱法对相关溶剂进行测定.因本品需要检测的有机溶剂的极性应与色谱柱的极性相似,所以采用中极性的6%氰丙基苯基-94%二甲基聚硅氧烷和交联聚乙二醇为固定相的毛细管柱为色谱柱,用FID检测器,选择以N,N-二甲基甲酰胺为溶剂,采用顶空法进行检测[6].本品中所使用的有机溶剂多为挥发性的物质,顶空进样的方式与直接进样的方式相比,可以减少色谱系统的污染.三乙胺属于强碱性溶剂,不易与其他溶剂同时进行检测,但仍采用相同的方法和条件.而4-二甲氨基吡啶较为特殊,其沸点较其他5种溶剂沸点更高,极性较大,所以单独采用交联聚乙二醇为固定相的毛细管柱作为色谱柱,采用直接进样的方式进行检测[7-8].
在帕瑞昔布合成及精制的过程中用到的溶剂有乙醇、二氯甲烷、乙酸乙酯、正己烷、4-二甲氨基吡啶、三乙胺,三乙胺的碱性很强,4-二甲氨基吡啶的沸点较高,所以在进行方法学验证的时候需要分开测定,才能对其准确地判断.乙醇、二氯甲烷、乙酸乙酯、正己烷的物理化学性质较为相似,因此,可把这4种溶剂放在一起测定.在采用气相色谱法测定时,首先先确定进样条件,选择最佳条件,采用顶空进样法;三乙胺的碱性很强,所以对三乙胺单独验证,条件与前面的条件基本相似,依然采用顶空进样;4-二甲氨基吡啶的沸点过高,所以采用直接进样法;最终确保了每一种溶剂的方法都是最合适的.实验结果显示,各检测项目均符合检测要求,残留量测定结果均符合药典中对于残留溶剂的规定.本方法操作简便、准确度高、分离效果好、灵敏度高、分析速度快,适合用于帕瑞昔布钠中残留溶剂的质量控制,进而确保帕瑞昔布钠产品的质量.
猜你喜欢
应用化工(2023年1期)2023-02-16 10:56:46
中华养生保健(2020年9期)2021-01-18 03:12:32
河南化工(2020年11期)2020-12-10 05:43:20
上海化工(2018年10期)2018-10-31 01:21:06
浙江化工(2018年3期)2018-04-19 08:42:56
癌症进展(2016年8期)2016-08-22 11:22:14
微生物学杂志(2015年1期)2015-12-26 08:18:01
化工生产与技术(2014年5期)2014-02-27 13:42:02
哈尔滨医药(2014年6期)2014-02-27 13:35:49
东南大学学报(自然科学版)(2012年3期)2012-06-28 03:59:00
