
肥厚性心肌病RAF1 c.770C>T基因突变1例并文献复习
2018-09-26 10:45
精准医学杂志 2018年4期
(四川大学华西第二医院儿童心血管科,四川 成都 610041)
肥厚性心肌病(HCM)是最常见的心肌病之一,可致儿童和年轻人心脏性猝死。通常,在无高血压、主动脉瓣狭窄等其他负荷条件时,HCM被定义为一种以心肌肥厚为特征的心肌原发性疾病,通常影响左心室功能[1]。心力衰竭或者心律失常是常见的主要症状,但也可能会经历一种良性和无症状的生理病理过程。到目前为止,HCM的诊断方法主要为超声心动图检查。然而,随着遗传和分子技术的迅速发展,发现了一系列HCM突变基因,这些突变基因主要和影响肌小节的功能有关[2],同时也发现了一些新的基因突变。本文报告1例年幼起病的HCM病儿,带有RAF1 c.770C>T突变,而未发现其他已知的临床综合征。现将结果报告如下。
1 临床资料
病儿,女,10个月,因发热、咳嗽5 d入我院治疗。病儿有吸奶间断伴呼吸困难,且自出生后反复发作性发绀,伴生长发育迟缓。入院体检发现心率168次/min,呼吸频率为72次/min,血压为11.70/5.85 kPa,下肢水肿,三凹征阳性,双肺呼吸音粗,肝脏右肋缘下4 cm,心尖区闻及收缩期杂音。动脉血气分析显示氧分压低于5.05 kPa。此外,心肌肌钙蛋白I 0.890 μg/L,脑利钠肽2 730.0 ng/L。病儿首诊为Ⅰ型呼吸衰竭、心功能不全和重症肺炎。治疗的同时行超声心动图及心脏大血管CT、MRI及心电图检查。超声心动图示左心室以及右心室心肌肥厚(图1A),左心室流出道狭窄伴血流速度加快,左心室收缩功能正常,但舒张功能不全。心脏大血管MRI检查亦显示左心室和右心室均肥厚(图1B),左心室射血分数为73.8%,收缩末期容积(ESV)为3.0 mL,舒张末期容积(EDV)为11.4 mL,右心室ESV为2.8 mL,EDV为8.6 mL(图1B)。心电图示左心室高电压。临床诊断HCM,给予抗生素控制感染,小剂量利尿剂减轻心脏负荷,并辅助呼吸机通气治疗,未给予正性肌力药物。治疗3 d后,病儿仍无法撤机,虽情况略有好转,但病儿父母因疾病可能预后不良而放弃治疗。
在父母知情同意下取病儿及其父母血样,提取DNA。使用BigDye3.1化学法(Applied Biosystems Foster City,CA,美国)对聚合酶链反应扩增子进行Sanger测序,并在ABI3130xl生物分析仪(Applied Biosystems Foster City,CA,美国)上进行毛细管电泳。所得测序色谱图与GenBank参考序列进行了比对,并筛选父母DNA标本以研究等位基因传递情况。在测序分析的基础上,发现病儿携带一种新的杂合突变体RAF1 c.770C> T(图2),但其父母未发现相同突变体。此外,在3个样品中确认了正常染色体核型的存在。
2 讨 论
HCM是一种常见的心肌病,临床症状及辅助检查曾是此类疾病最重要的诊断依据。然而,从20世纪90年至今,人们对HCM的遗传和分子机制研究取得了很大进展。已有的分子遗传学研究对编码肌小节蛋白或者肌小节相关蛋白等基因进行筛查,发现HCM病人携带的突变基因集中在MYH7、MYBPC3、TNNT2、TNNI3、TPM1、ACTC、MYL2和MYL3等8个基因[1-3]。这些致病相关基因所表达的蛋白质,主要参与肌小节形成,由这些突变基因表达出的功能缺陷蛋白组装入肌小节,可导致心肌纤维结构与功能异常[4]。此外,有研究还进行了细胞和转基因动物模型的功能实验,以展示类似的病理生理过程,为心肌病的遗传和分子机制研究提供了重要依据[5-7]。
本研究发现了RAF1 c.770C>T新突变体,根据HCM诊断和治疗指南[8],RAF1不属于肌小节相关调节基因。因此,RAF1 c.770C>T突变被认为是一种潜在的HCM DNA变异,这种变异可能是HCM的病因之一。此外,研究发现RAF1突变可影响RAF1激酶的活性,可能导致具有HCM特征的NOONAN和LEOPARD综合征的发生[9]。为排除这两种综合征,本研究对样本再次行染色质分析,结合病儿无任何特殊面容及无大小不同褐色斑点,亦无瓣膜发育异常、骨骼畸形、心脏传导障碍和耳聋等相关症状,排除了这两种综合征的可能性。
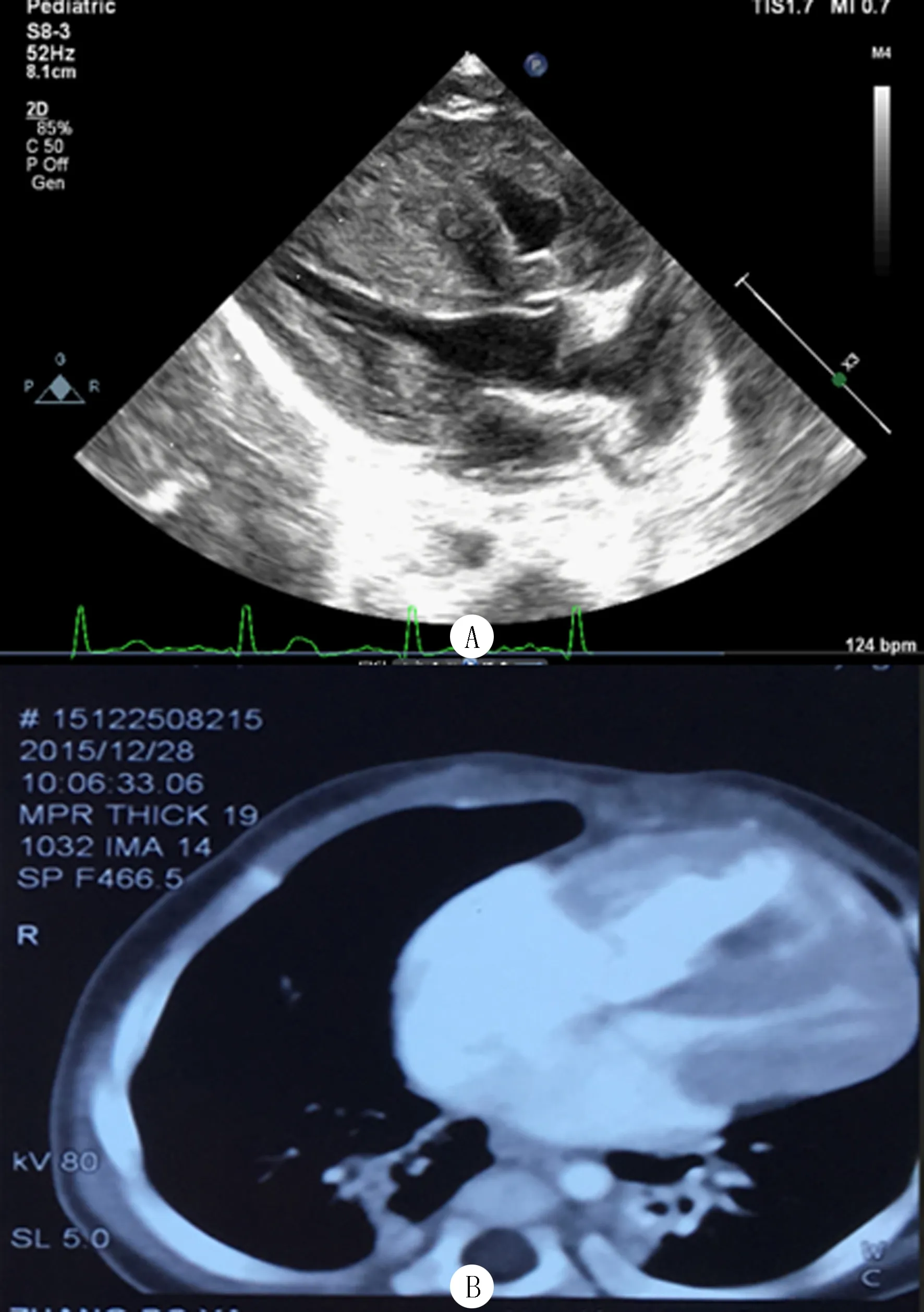
A:超声心动图;B:心脏MRI。
图1HCM病儿超声和心脏MRI检查表现
病儿发生RAF1 c.770C>T基因突变,而病儿父母无RAF1基因突变。
图2HCM病儿及其父母基因测序结果
根据基因测序诊断步骤[10],本研究未发现病儿父母具有相同的突变。目前对HCM病人的管理、治疗和预后主要是根据临床和超声心动图等检查结果,而不是基于遗传背景。所以,目前存在这样一个问题,尽管基因检测在HCM中很重要,基础研究已经取得了很大的进步,但该病儿突变结果在临床实践中的实用性是有一定的局限性的,无法对该病儿进行更为精准的管理和治疗。后续随访家长得知,病儿出院10 d后死亡。
相关研究结果提示HCM伴明显右室肥厚者比不伴右室肥厚者的心血管不良事件大大增加,预后欠佳[10],同时RAF1基因与扩张型心肌病相关[11]。有证据表明,RAF1编码下游RAS效应子,并通过NFAT诱导肥厚信号传导[9,12]。如上所述,我们考虑RAF1 c.770C>T的非肌小节相关突变可能是该病儿早发HCM的遗传原因,该突变可能在病儿年龄较小时即可诱发明显的心室肥大,尤其是导致右心室和流出道梗阻,有可能在婴儿期出现心力衰竭和猝死,这应该是与其他肌小节相关基因突变引起HCM的主要区别。
目前与HCM相关的基因突变多与肌小节蛋白相关,所得到突变基因也与其他疾病相关,多个发现的候选基因也是扩张性心肌病及左心室心肌致密化不全的疾病相关基因。相关基因突变信息大多来自于候选基因筛查,覆盖范围较窄,而且易受其他疾病表型的影响,因此,研究人员在接近HCM病因真相方面似乎仍有很长的路要走。到目前为止,我们仍在努力寻找途径尝试通过基因筛查发现可能的基因突变,并期望根据突变结果对处于心室肥厚进展的病人进行临床前诊断,然后选择恰当的干预方式,但收效有限。目前可能所能做的就是及时更新指南:比如基于遗传检测结果如何确定疾病的风险阶段和预后;应进行大样本的观察研究,确定特定基因型是否与生存率相关;是否应根据个体基因组测序而执行单独的治疗策略,而非仅仅基于其临床和超声心动图表现;以及研发治疗HCM和其他心肌病需要的靶向药物等,这些应该是基因研究的最终目的。
猜你喜欢
安徽医药(2021年4期)2021-04-09
中国生殖健康(2020年2期)2021-01-18
安徽医药(2020年4期)2020-04-16
安徽医药(2020年1期)2020-01-10
中国临床医学影像杂志(2019年1期)2019-04-25
精准医学杂志(2018年5期)2018-11-16
小学生导刊(2018年13期)2018-06-29
中国生殖健康(2018年2期)2018-01-12
中国当代医药(2015年31期)2015-03-01
中国医药科学(2015年15期)2015-02-27
