
基于氧化石墨烯的环糊精型色谱固定相的制备及在对映体分离与亲水色谱中的应用
2018-09-11 12:03李媛媛高柱仙李添君马玉龙
分析化学 2018年9期
李 强 李媛媛* 朱 楠 高柱仙 李添君 周 彤 马玉龙
1(宁夏大学省部共建煤炭高效利用与绿色化工国家重点实验室, 银川 750021)2(宁夏大学化学化工学院, 银川 750021)
1 引 言
手性药物对映体通常在药理活性、代谢、稳定性和毒性等方面具有显著性差异,当手性药物作为外消旋混合物给药时,一种对映体具有治疗所需的药理学作用,而另一种对映异构体不具有作用甚至有害[1]。由于手性药物对于生物体的强烈作用以及防止手性药物作为外消旋混合物直接销售,手性药物的有害对映体部分必须被彻底分离[2]。因此对映体分离在制药领域具有重要意义[3,4],是分析化学领域的研究热点。液相色谱是最常用的对映体拆分方法之一,目前手性色谱固定相根据其化学类型主要包括刷型[5]、环糊精(CD)及其衍生物[6,7]、大环抗生素[8]、纤维素[9]、蛋白质[10]和金属有机骨架(MOF)[11,12]等。
石墨烯(G)由于具有超高的比表面积和高度离域π电子共轭体系,对芳香族化合物以及其它具有共轭结构的化合物表现出较强的吸附作用和较高的吸附容量[13~16]。氧化石墨烯(GO)不仅含有丰富的含氧官能团,而且它的大离域电子系统可以为碳环结构提供强大的亲和力,碳环结构在药物、污染物和生物分子中的应用广泛。通过共价键合的方式制备GO修饰的色谱柱中,GO极大地提高了固定相的比表面积、改善了所制备色谱柱的分离性能,π-π堆积作用、疏水作用和氢键作用等使得烷基苯、中性多环芳烃、苯酚类化合物、苯胺类化合物以及核酸碱基等均得到良好分离效果[17,18]。β-环糊精(β-CD)是具有独特疏水性空腔的环状低聚糖,具有大量分子的稳定包合物的优异能力[19,20]。β-CD进行手性拆分主要依赖于内部空腔对分析物的包合作用和功能基团与分析物之间的相互作用。β-CD的包合作用、空腔大小和分析物结构密切相关,当分析物的疏水基团与空腔匹配较好时,对映体选择性较好,手性识别能力也较高[21~23]。将(3-巯基丙基)三甲氧基硅烷-β-环糊精(SH-β-CD)作为手性选择剂共价键合到SiO2@Au表面,制备基于SiO2@Au-SH-β-CD的色谱柱用于手性分离,实现了多种结构不同对映体的分离[24]。
亲水作用色谱(HILIC)是一种以极性固定相(如硅胶、氨基键合硅胶等)及含高浓度极性有机溶剂和低浓度水溶液为流动相的色谱模式。作为对反相色谱的补充和正相色谱的替代,HILIC已经显示出巨大的优势和潜力,成为分析极性和亲水性化合物的一种强有力的色谱技术[25~28]。
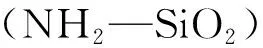
2 实验部分
2.1 仪器与试剂
TENSOR-27型傅里叶变换红外光谱(FT-IR)仪(德国布鲁克公司); HT7700型透射电子显微镜(TEM,日本日立高新技术公司); Vario EL cube型元素分析仪(德国Elementar公司); SETARAM SETSYS16型综合热分析仪(法国塞塔拉姆公司),以10℃/min的升温速度从室温至700℃; JSM-7500F型场发射扫描电子显微镜(SEM, 日本电子株式会社); FL2200-2型高效液相色谱仪(浙江福立分析仪器有限公司)。
硅胶(日本富士硅化学株式会社, 5 μm, 孔径120Å); GO(湖南丰化材料发展有限公司); 甲醇(色谱级)、乙腈(色谱级)、N,N-二甲基甲酰胺(DMF)、二氯亚砜(SOCl2)、甲苯、乙醇、丙酮、苯甲酸和苯甲醇(天津大茂化学试剂厂);β-CD、3-氨丙基三甲氧基硅烷(APTMS)、N,N-二环己基碳二亚胺(DCC)(阿法埃莎(中国)有限公司); 1-苯基乙醇、盐酸普萘洛尔、安息香、布洛芬、吡喹酮、R,S-雌马酚、酪氨酸、胸腺嘧啶、肌苷、鸟苷和腺苷(上海阿拉丁生化科技股份有限公司)。除特殊标明外,其它试剂均为分析纯。
2.2 实验方法
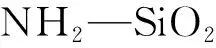
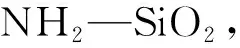
2.2.3β-CD-GO@SiO2的制备将2.38 g GO@SiO2和20 mL SOCl2混合, 75℃下加热回流10 h,加入2.00 gβ-CD和30 mL DMF,在N2保护下加热回流40 h。用DMF、蒸馏水洗涤3次后,干燥,得到手性固定相。β-CD-GO@SiO2固定相制备示意图如图1所示。
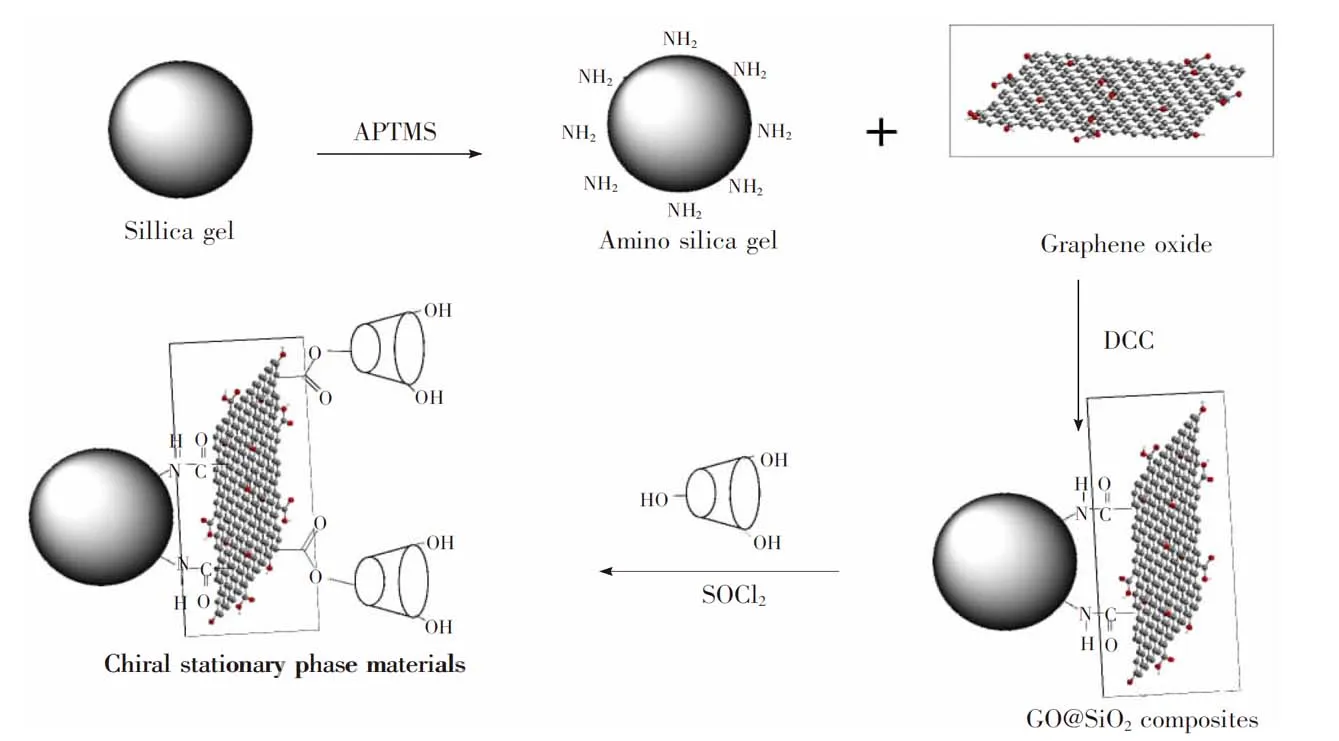
图1 β-CD-GO@SiO2固定相制备流程图Fig.1 Schematic diagram for preparation of β-CD-GO@SiO2 stationary phase. β-CD, β-Cyclodextrin; GO, graphene oxide; APTMS, aminopropyltrimethoxysilane; DCC, dicyclohexylcarbodiimide
2.3 色谱条件
将β-CD-GO@SiO2在60 MPa压力下用纯甲醇作为匀浆液和顶替液装入不锈钢管(150 mm×4.6 mm i.d.)中。所有色谱分离均在配有UV-vis检测器的FL2200-2型HPLC系统上进行。流动相流速为1 mL/min, 柱温为25℃。 根据公式(1)计算保留因子(k′)。
(1)
其中,t0是1,3,5-三叔丁基苯在β-CD-GO@SiO2柱上的保留时间(2.024 min),t是柱上对映体的保留时间。
根据公式(2)计算选择性因子(α)。
(2)
3 结果与讨论
3.1 β-CD- GO@SiO2材料表征

表1 元素分析结果
Table 1 Elemental analysis results
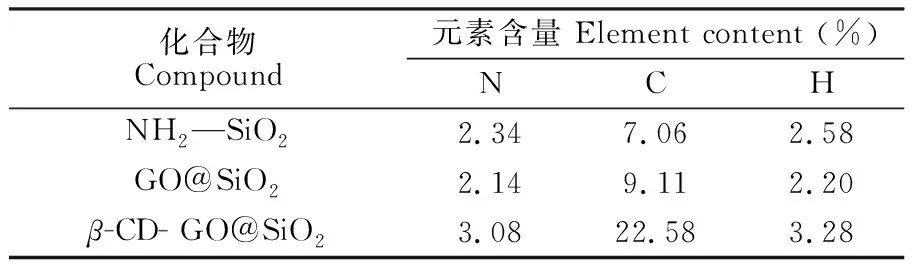
化合物Compound元素含量 Element content (%)NCHNH2SiO22.347.062.58GO@SiO22.149.112.20β-CD- GO@SiO23.0822.583.28
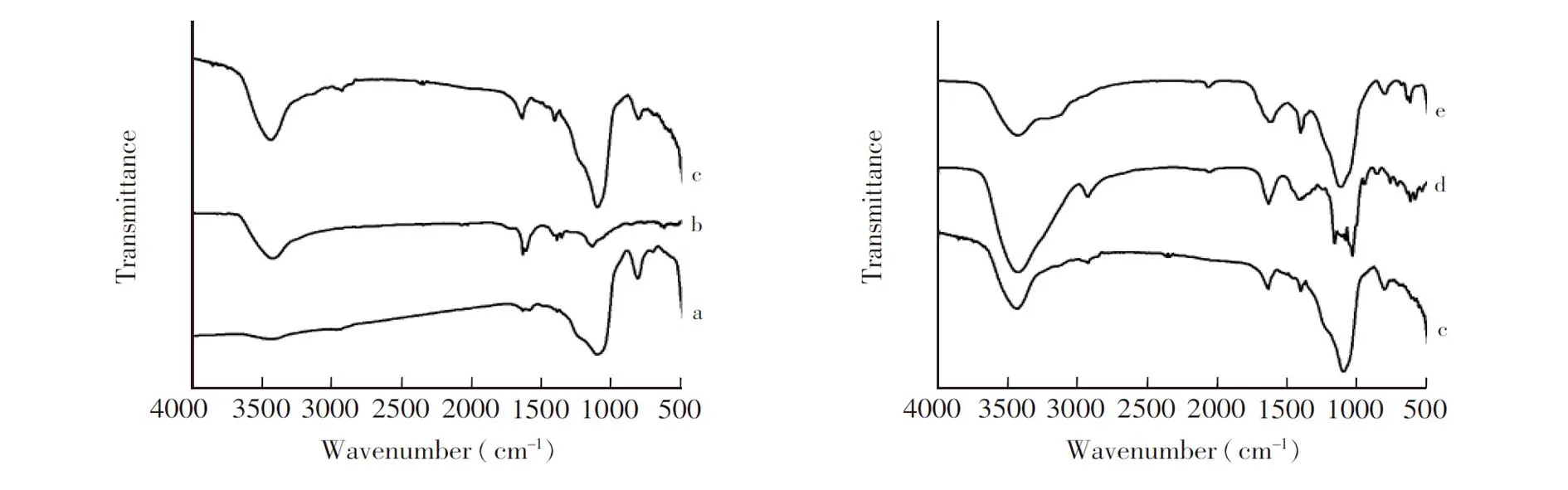
图2 红外光谱图: (a) NH2-SiO2,(b)GO,(c) GO@SiO2,(d)β-CD和(e) β-CD-GO@SiO2Fig.2 Fourier transform-infrared (FT-IR) spectra of (a), (b) GO, (c) GO@SiO2, (d) β-CD and (e) β-CD-GO@SiO2

图3 GO@SiO2的扫描电镜图(A)和透射电镜图(C); β-CD-GO@SiO2的扫描电镜图(B)和透射电镜图(D)Fig.3 Scanning electron microscopy (SEM) (A) and transmission electron microscopy (TEM) (C) images of GO@SiO2; SEM (B) and TEM (D) images of β-CD-GO@SiO2
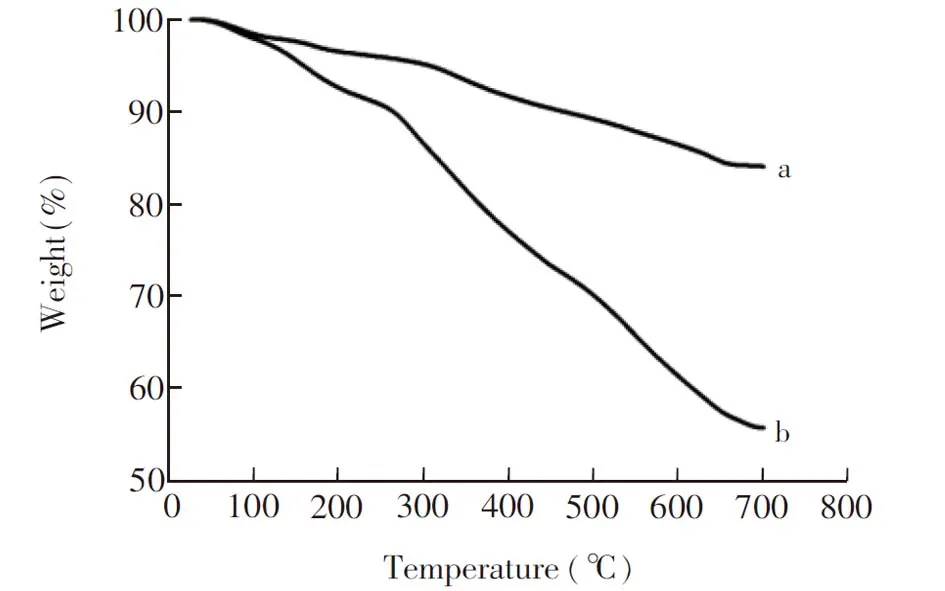
图4 (a) GO@SiO2 和 (b) β-CD- GO@SiO2的热重曲线Fig.4 Thermogravity curve of (a) GO@SiO2 and (b) β-CD-GO@SiO2
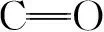
3.2 β-CD-GO@SiO2固定相在手性分离中的应用
3.2.17种手性对映体物质的分离β-CD-GO@SiO2色谱柱分离4种典型对映体的色谱图如图5所示。用k2、α和分离度(Rs)作为参数来评价该固定相的分离情况,如表2所示。由分离数据可知,7种手性物质得到不同程度的分离,并且对映体在固定相上的保留强,分离度未必高,对映体的结构对手性分离有很大影响。1-苯基乙醇的Rs>2,可能是因为1-苯基乙醇空间位阻较小。含苯环部分相对疏水容易进入空腔内部而极性较大的含手性部分处于空腔两侧[4,30],手性中心与β-CD匹配较好,分离效果更为明显。安息香、R,S-雌马酚和普萘洛尔在固定相上的Rs>1.5,从结构上看,它们都有多个苯环和羟基,一方面可增强GO上共轭体系与苯环化合物的π-π堆积作用,有利于靠近β-CD,并容易进入β-CD内腔产生包合作用,促进分离; 另一方面,GO和β-CD上的羧基与羟基可与分析物形成氢键作用,使得Rs增加。与未键合GO的固定相研究相比[31,32],键合GO在一定程度上能提高对映体分离能力,GO参与并促进了手性分离。
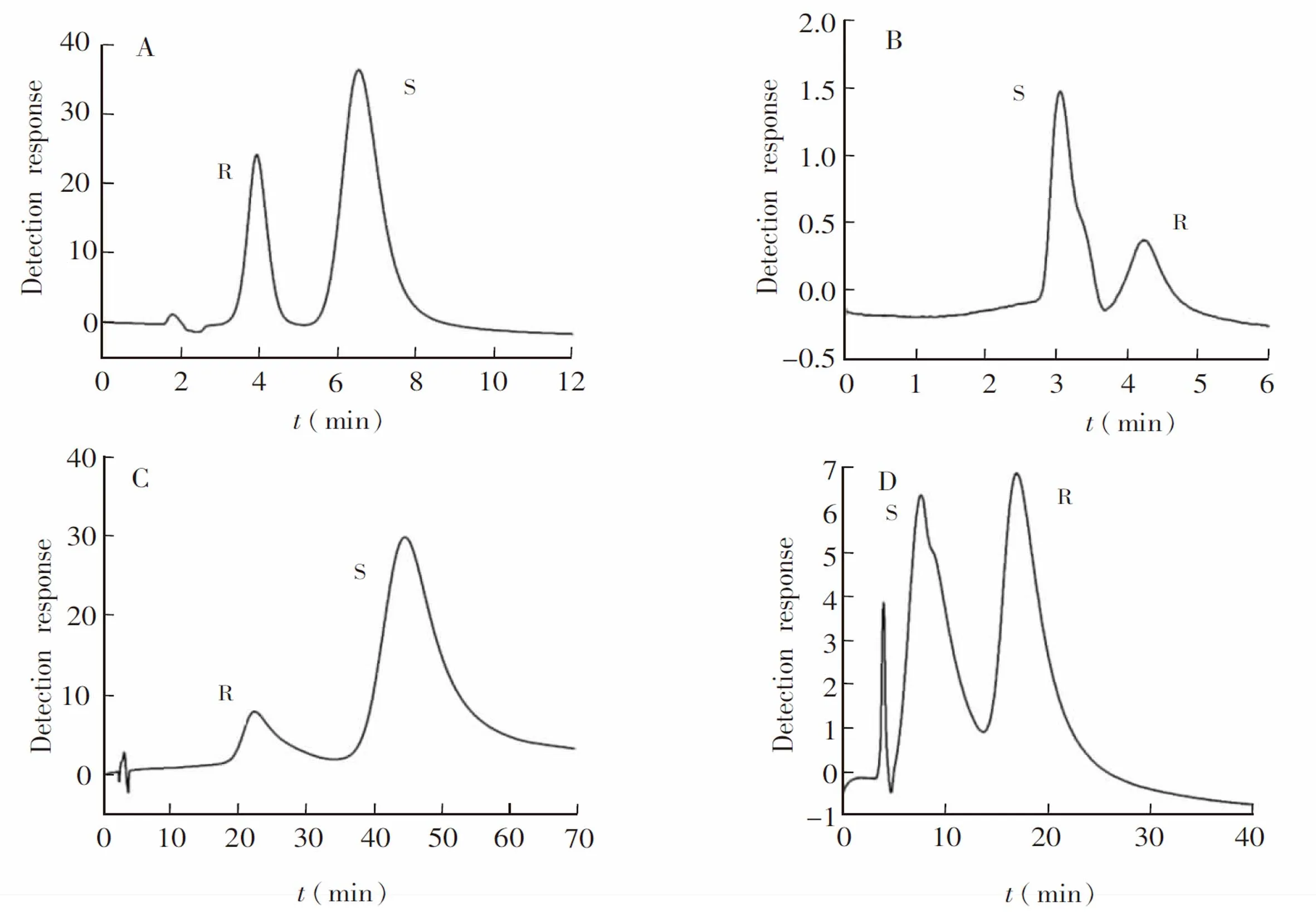
图5 4种典型手性化合物的色谱图:(A)1-苯基乙醇,(B)普萘洛尔,(C)安息香,(D)酪氨酸。流动相比例及检测波长同表2Fig.5 Chromatograms of four typical chiral compounds: (A) 1-phenylethanol, (B) propranolol, (C) benzoin and (D) tyrosine. The mobile phase and detection wavelength are the same as in Table 2
表2 手性化合物分离数据
Table 2 Separation data for chiral compounds
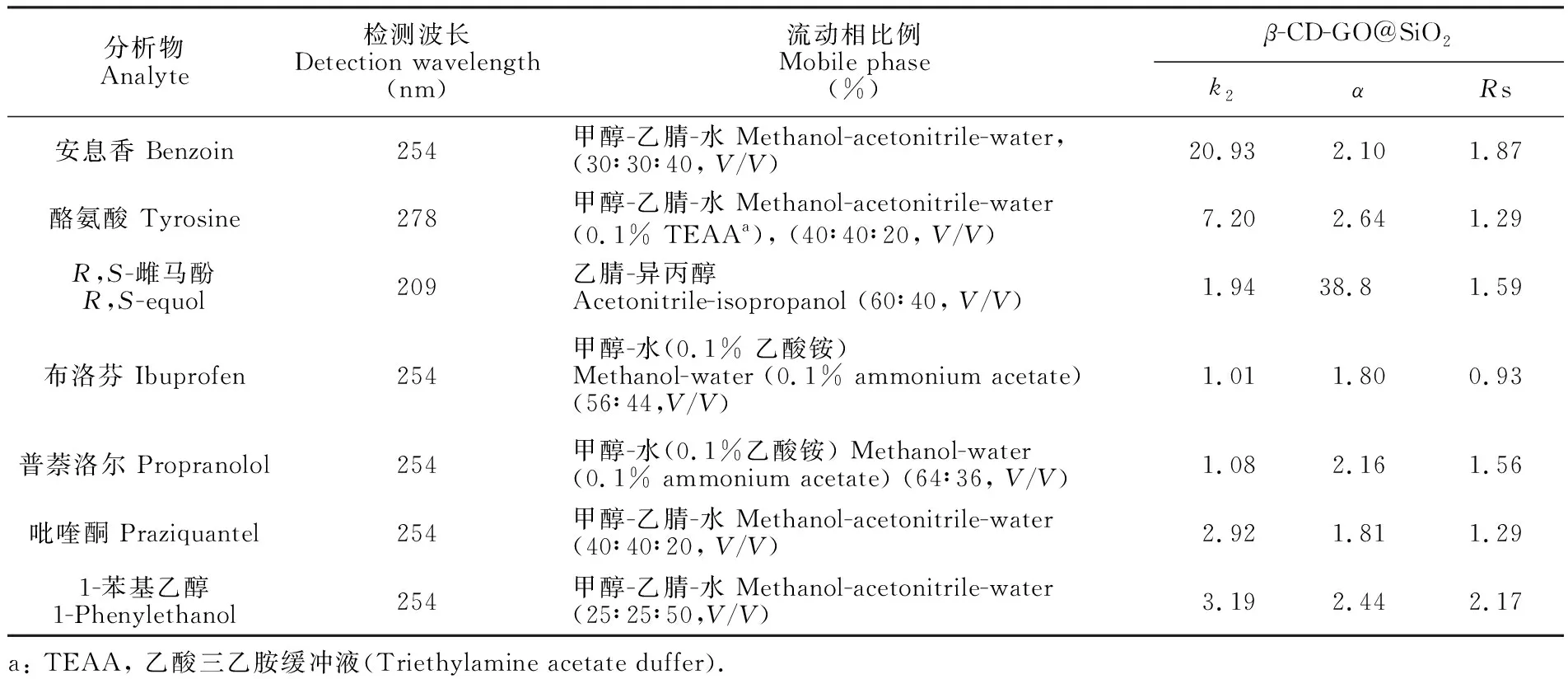
分析物Analyte检测波长Detection wavelength(nm)流动相比例Mobile phase(%) β-CD-GO@SiO2k2αRs安息香 Benzoin254甲醇-乙腈-水 Methanol-acetonitrile-water,(30∶30∶40, V/V)20.932.101.87酪氨酸 Tyrosine278甲醇-乙腈-水 Methanol-acetonitrile-water(0.1% TEAAa), (40∶40∶20, V/V)7.202.641.29R,S-雌马酚R,S-equol209乙腈-异丙醇Acetonitrile-isopropanol (60∶40, V/V)1.9438.81.59布洛芬 Ibuprofen254甲醇-水(0.1% 乙酸铵)Methanol-water (0.1% ammonium acetate)(56∶44,V/V)1.011.800.93普萘洛尔 Propranolol254甲醇-水(0.1%乙酸铵) Methanol-water(0.1% ammonium acetate) (64∶36, V/V)1.082.161.56吡喹酮 Praziquantel254甲醇-乙腈-水 Methanol-acetonitrile-water(40∶40∶20, V/V)2.921.811.291-苯基乙醇1-Phenylethanol254甲醇-乙腈-水 Methanol-acetonitrile-water(25∶25∶50,V/V)3.192.442.17a: TEAA, 乙酸三乙胺缓冲液(Triethylamine acetate duffer).
3.2.2温度对手性分离的影响对映体拆分热力学行为的研究是深入了解手性识别机理和色谱柱稳定性的有效手段。如下Van′t Hoff equation公式[33]显示了分析物从流动相转移到固定相的焓变(ΔH)和熵变(ΔS)。其中R,T和Ф分别是气体常数、绝对温度和相比。
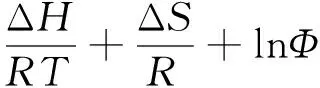
(3)
在采用高效液相色谱分离对映体的研究中,大多数分析物在填充柱上的保留和选择性受柱温影响,且溶质与β-CD的结合常数受温度的影响也很大。在低温下,结合常数增大,但是传质递变会变差,所以降低柱温可使β-CD键合相的选择性增大,同时也可能使分离度改善。在20.0~60.0℃的温度范围内对1-苯基乙醇在手性柱上的拆分情况进行探究,分离色谱图如图6A所示。随着柱温升高,两种1-苯基乙醇对映异构体的保留时间减少,导致k′、α和Rs值降低, 说明1-苯基乙醇的分离过程是放热的。图6B中的对映体Van′t Hoff equation曲线表明,1-苯基乙醇分离线性关系良好(R2>0.996),说明手性柱的构象和对映选择性机制与温度无关。从lnk′-1/T直线的斜率和截距得到ΔH和ΔS(表3)。随温度升高,1-苯基乙醇保留时间减小,说明温度对两个对映体的作用机理相似,都是焓值驱动原理。
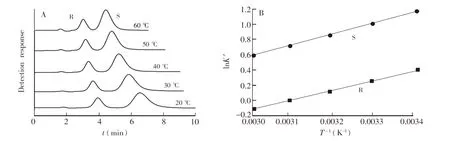
图6 (A) 1-苯基乙醇在β-CD-GO@SiO2上的色谱分离图,使用甲醇-乙腈-水(27.5∶27.5∶45, V/V)作为流动相,在紫外波长254 nm处检测; (B) 在β-CD-GO@SiO2上分离1-苯基乙醇的lnk′-1/T图Fig.6 (A) Separation chromatograms of 1-phenylethanol on β-CD-GO@SiO2, methanol-acetonitrile-water (27.5∶27.5∶45, V/V) as the mobile phase with UV detection at 254 nm; (B) lnk′-1/T plot for separation of 1-phenylethanol on β-CD-GO@SiO2
3.3 β-CD-GO@SiO2固定相在亲水色谱中的应用
3.3.1流动相和柱温对分析物保留的影响在HILIC中,流动相是影响极性及亲水性分析物保留的最主要的因素。分别选择乙腈和乙腈/甲醇作为有机相考察流动相对4种核苷小分子保留的影响。如图7A和7C所示,当有机相含量从50%增加到80%时,4种分析物的保留逐渐增加,当有机相含量增加至90%或以上时, 分析物保留急剧增加,这是典型的HILIC保留特征。对比图7A和7C发现,随着有机改性剂甲醇的加入,4种分析物的保留显著减弱,这是由于有机改性剂甲醇是质子化溶剂,与溶质分子相互竞争与β-CD和GO作用,使分析物与固定相之间的作用减弱,洗脱能力增强。但有机改性剂甲醇的加入,使得材料的亲水作用增强,对于4种分析物的分离具有明显的促进作用。
表3 ΔH, ΔS, ΔG(25℃), andR2的值
Table 3 Values of ΔH, ΔS, ΔG(25℃) andR2
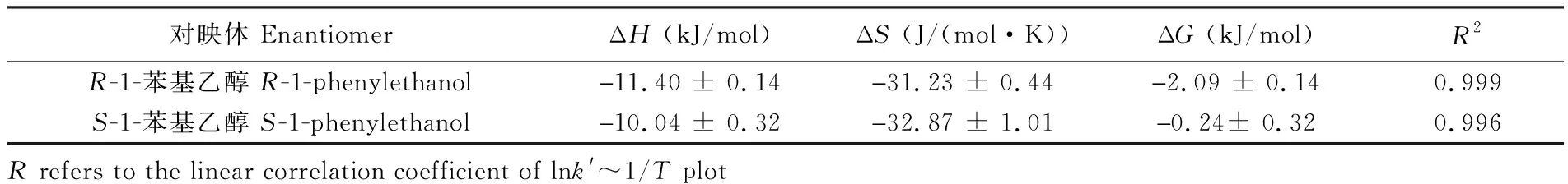
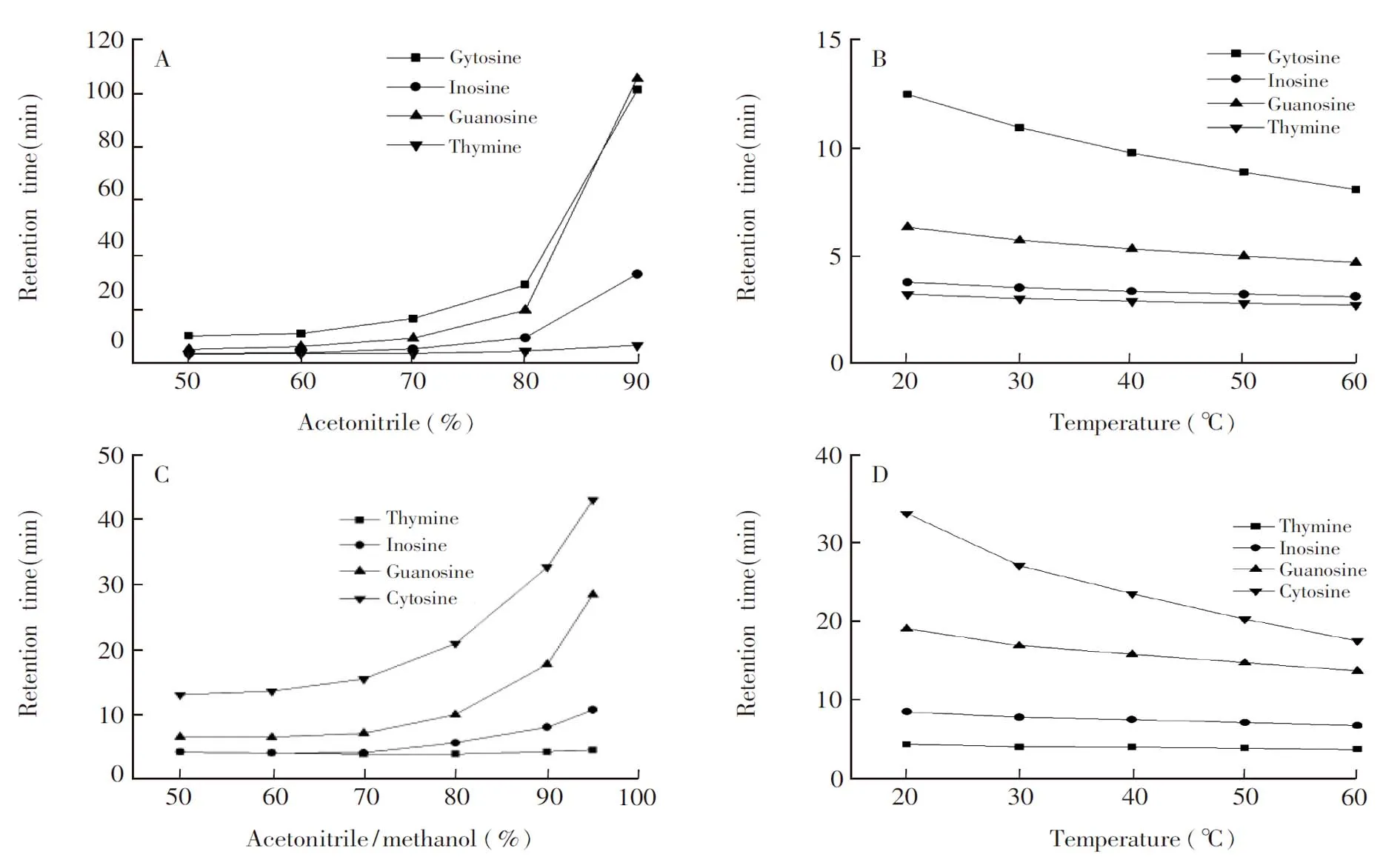
图7 不同流动相和温度对分析物保留的影响:(A)流动相为乙腈-水; (B)流动相为乙腈-水(90∶10, V/V); (C)流动相为乙腈-甲醇-水; (D)流动相为乙腈-甲醇-水(45∶45∶10, V/V)Fig.7 Effect of different mobile phase and temperature on the retention: (A) acetonitrile-water; (B)acetonitrile-water (90∶10, V/V); (C) acetonitrile-methanol-water; (D) acetonitrile-methanol-water (45∶45∶10, V/V)
在HILIC中,柱温的改变可以影响扩散系数、流动相的粘度和分析物在流动相和固定相之间的转移焓变,从而影响分析物的保留。以上述4种核苷分子为分析物,分别以乙腈-水(90∶10,V/V)和乙腈-甲醇-水(45∶45∶10,V/V)为流动相,通过改变温度(20~60℃)研究温度对核苷类物质保留的影响。如图7B和7D所示,所有分析物的保留时间均随着柱温增加而减小。由于保留时间在很大程度上取决于柱温,当柱温增加时,分析物在流动相和固定相表面富集水层之间的分配系数发生变化,从而导致保留时间减小,说明分析物在色谱柱的保留行为是放热过程。
3.3.2流动相pH值对分析物保留的影响流动相pH值也是影响分析物保留的因素之一。选择苯甲酸、布洛芬、腺苷、胞嘧啶、苯甲醇作为目标分析物,考察它们在pH为2.8~6.3之间的保留情况。如图8A所示, pH值在2.8~5.3时,苯甲酸和布洛芬的保留值随着流动相pH值的增大而增大,由于它们的pKa分别为4.2和4.6,当pH值从2.8增加到5.3时,苯甲酸和布洛芬由分子状态逐渐离解,与固定相之间的静电作用增强,导致保留增加,当pH值继续增加到6.3时,苯甲酸和布洛芬完全离解,离子状态不发生变化,故而保留值不发生显著变化。胞嘧啶和腺苷在研究的pH值范围内,保留值随着流动相pH值的减小而增大。这是由于腺苷和胞嘧啶是碱性物质,当pH值从6.3降低到2.8时,它们逐渐质子化,与固定相之间静电吸引作用增强导致保留增强。pH值的变化对苯甲醇的保留影响不大。
图8 (A) 流动相pH对分析物保留的影响; (B) 4种极性生物分子在HILIC模式下的色谱分离图Fig.8 (A) Effect of mobile phase pH on the retention; (B)Separation chromatogram of four polar biomolecules on hydrophilic interaction chromatography (HILIC)
3.3.34种核苷小分子的分离图8B是流动相为甲醇-乙腈-水 (45∶45∶10,V/V)、温度为25℃时,4种生物分子的色谱分离图,在此条件下,4种分析物实现了基线分离。由分子式可知,胞嘧啶和胸腺嘧啶分子结构相似,但胞嘧啶多1个氨基,与β-CD形成氢键作用,因此吸附作用增强但解析时间增加,导致保留时间变长且峰形变宽; 而胸腺嘧啶相对于胞嘧啶,与β-CD结合力弱,容易被洗脱,因而保留时间短而峰形窄。肌苷与鸟苷的分子式结构相似,而鸟苷多1个氨基,因而肌苷更容易洗脱。分析物按照胸腺嘧啶、肌苷、鸟苷、胞嘧啶的顺序依次被洗脱,按照HILIC机理,溶质按极性增大的次序依此被洗脱。由于溶质在色谱上的保留是十分复杂的过程,既有在有机相和富水层之间的分配,也有氢键、偶极作用和静电作用等一些复杂作用。因此,分析物的洗脱顺序与其分子结构密切相关。
4 结 论
本研究成功制备了β-CD-GO@SiO2型固定相并应用于7种手性对映体的拆分,结果表明,GO和β-CD在手性识别过程中具有协同作用,氢键、π-π堆积作用在手性识别中具有改善分离的作用。此外,此固定相还可以作为亲水色谱固定相,用于极性和亲水性小分子的分离。这种固定相实现了一根柱子两种分离模式,使用灵活,并且可以扩大分析样品的范围,具有良好的应用潜力。
猜你喜欢
井冈山大学学报(自然科学版)(2021年3期)2021-09-10
吉林大学学报(理学版)(2021年3期)2021-05-26
农药科学与管理(2019年8期)2019-11-23
中成药(2017年9期)2017-12-19
百科知识(2015年13期)2015-09-10
质谱学报(2015年5期)2015-03-01
医药导报(2015年6期)2015-02-10
化学工业与工程(2015年1期)2015-02-10
无机化学学报(2014年12期)2014-02-28
无机化学学报(2014年7期)2014-02-28
