
低维原子/分子晶体材料的可控生长、物性调控和原理性应用∗
2018-07-10 09:32黄立1李更1张余洋2鲍丽宏1郇庆1林晓2王业亮1郭海明1申承民1杜世萱1高鸿钧1
物理学报 2018年12期
黄立1)2) 李更1)2) 张余洋2)1) 鲍丽宏1)2) 郇庆1)2) 林晓2)1)王业亮1)2) 郭海明1)2) 申承民1)2) 杜世萱1)2) 高鸿钧1)2)†
1)(中国科学院物理研究所,纳米物理与器件实验室,北京 100190)
2)(中国科学院大学物理科学学院,北京 100049)
(2018年4月28日收到;2018年5月8日收到修改稿)
1 引 言
低维纳米功能材料是多种新奇物理现象和物理问题的载体,是电子器件实现纳米化和量子化的基础和核心,是学术界和工业界的重要研究方向[1−5].在近年来的研究中,科学家们在原子/分子操纵、单分子器件、单分子性能调控、精确构筑图案化纳米结构和基于第一性原理的理论计算等方面取得了一系列重大突破,解决了一些低维纳米功能材料的基本物理问题,并为其实用化奠定了基础.在众多低维纳米材料中,石墨烯得益于其单原子层结构,具有许多新奇的物理性质,掀起了新型二维原子晶体材料的研究热潮,各种新材料和新结构被构筑,研究人员对与之相关的物理问题也开展了深入的研究.
在低维纳米功能材料的研究中,人们普遍关心的问题包括:1)低维纳米功能材料的可控生长与机制研究;2)低维纳米功能材料在超高密度信息存储方面的应用;3)纳米量子结构自旋态的调控;4)晶圆尺寸石墨烯及新型二维原子晶体材料的可控构筑及物性调控;5)纳米级精准图案化量子结构的构筑方法;6)新型纳米功能材料在电子和光电器件上的应用等.
高鸿钧课题组在物理所20多年来的研究中,聚焦于低维纳米功能材料的可控生长、机制与物性,系统地研究了固体表面的有机分子和以石墨烯为代表的二维原子晶体两大类材料,取得了一系列研究成果,本文介绍了其中部分代表性工作.
2 几种低维纳米量子结构的可控生长与机制研究
低维纳米量子结构的可控生长是制备器件和优化器件性能的基础.我们的研究聚焦于原子和有机分子在单晶表面的吸附、组装与功能化,借助扫描隧道显微学表征和第一性原理计算研究其物理本质,进而揭示可控生长的机制.
2.1 纳米尺度“海马”分形结构的形成及其生长机制
自然界存在着许多令人惊叹的美丽的分形图案,比如大树的枝杈、叶脉和花序等.在物理学上用分形结构来描述这类相似现象,在数学上也有专门研究自相似结构的几何学,即分形学.对于分形结构的生长动力学,一般可以用扩散限制凝聚(di ff usion limited aggregation,DLA)模型和团簇扩散限制凝聚(cluster di ff usion limited aggregation,CDLA)模型来描述.
我们系统地研究了富勒烯聚合物薄膜分形结构的生长.富勒烯是一种在电学、光学和光电等方面具有优异特性的有机材料[6−8].我们利用离子团束沉积方法,制备出了C60-TCNQ(Tetracyanoquinodimethane,四氰基对苯二醌二甲烷)和纯TCNQ有机薄膜,并第一次在该薄膜中观察到一种奇特的准二维分形结构[6,7,9],形似“海马”,呈现S形,即具有弯曲的主干,其边缘长有小的“鳍”.每个S形的图案都明显地与二维反转对称不同,这种对称性破缺在当时薄膜生长的文献中非常少见.样品的透射电子显微镜(transmission electron microscopy,TEM)明场像显示,在薄膜的大部分区域有很多团簇,少部分没有团簇的区域出现了这种“海马”分形结构.在没有团簇的区域外侧,团簇的密度达到峰值.图1(a)显示了海马结构的细节,分形结构沿海马的主干方向生长,在海马结构附近没有团簇,这说明海马是这一区域的团簇凝聚而成的.图1(b)展示了由这些海马结构组成的三行平等排列的分形结构.在这三行结构中,每一个海马结构几乎是完全相同的,显示出高度自相似的特性.
这一分形结构的生长机制可以用CDLA模型解释.首先,分散的团簇组成分形结构的核,并按S形排列在一起;然后,这些核扩大并连接在一起,但是沿着S形方向的生长速率要大于垂直于S形的方向,即出现CDLA;最后,这些核生长成为海马状分形结构.根据实验结果,我们提出了一种全新的生长机制:在成核生长过程中,由于存在部分带电团簇,使得临界核荷电因受库仑排斥作用而导致对称性破缺,最终形成“海马”分形结构[10,11].

图1 在C60-TCNQ多层薄膜中观察到的分形结构的TEM图像[6](a)海马分形结构的微观细节;(b)低倍率TEM图像显示三行平行排列的分形结构Fig.1.TEM images of the fractal pattern observed in C60-TCNQ multilayered thin fi lm[6]:(a)TEM image showing the detailed structure of the seahorse-like fractal structures;(b)low-magni fi cation TEM image showing three rows of the separated fractal patterns which are almost parallel to each other and possess the self-similarity property.
目前,没有观察到任何其他材料可以生长出这种“海马”图案.理解这种新颖形貌的形成机制是一个挑战.特别是这种二维的手性“海马”图案不存在任何转动关联,而仅仅是镜面反演,因此在图案形成过程中自发地存在二维反演对称性破缺.我们提出的生长机制认为,在这类分形结构中,二维的反演对称性只是在整个宏观尺度上被破坏,荷电效应、分子极化和诱导偶极或四极相互作用等在图案的形成过程中起了重要作用.
2.2 扫描隧道显微镜分辨率的提高及最高分辨Si(111)-7×7原子图像的获得
半导体硅(Si)表面结构及锗(Ge)在Si表面上生长的研究是21世纪初的重要研究课题之一.其中,对Si(111)-7×7表面上顶戴原子(adatom)和静止原子(rest atom)的直接观察,是自1981年扫描隧道显微镜(scanning tunneling microscopy,STM)出现以来人们所追求的目标之一.然而,在最初的20年里,只有IBM的Avouris研究组给出了Si(111)-7×7单胞中12个adatom和无层错半单胞(unfaulted half unit cell)中的3个rest atom的STM图像,而在有层错单胞中未能显示任何rest atom[12].Giessibl[13]用另一种手段——低温超高真空原子力显微镜(ultra-high vacuum lowtemperature atomic force microscopy,UHV LTAFM),观察到了该单胞中所有6个rest atom和12个adatom,其研究成果发表在《Science》上.然而,其中的rest atom很难清楚地分辨出来.
我们利用室温UHV-STM对Si(111)-7×7表面结构进行了深入系统的研究.通过改进STM针尖,在国际上首次用STM同时清晰地分辨出单胞中的所有6个rest atom和12个adatom,并且rest atom的亮度与无层错半单胞内中心adatom的亮度相同.第一性原理计算表明,该高分辨STM图像的获得与STM针尖曲率半径和扫描偏压有关.如图2所示,利用针尖半径为7 Å的针尖在合适的偏压下(如−1.5 V)计算模拟所得到的STM图像能同时分辨单胞中的所有6个rest atom和12个adatom,与STM实验结果完全符合[14].
该成果获得了自STM发明20年以来最高分辨的Si(111)-7×7的STM图像,同时提出了针尖尖端的大小是决定STM是否能获得表面纳米结构中更加精细的电子结构信息的关键.进一步的研究表明,通过对STM针尖的修饰,可以获得表面功能有机分子的精细的电子结构信息[15−17].这些工作对纳米和表面科学领域具有十分重要意义.
在清晰分辨Si(111)-7×7表面结构的基础上,我们对功能结构在其表面的吸附行为开展了研究.对Si(111)-7×7表面上Ge的初期吸附位置的研究,是Ge/Si体系中的重要研究内容,它决定所生长体系的结构与物理性质.但是,在21世纪初,尽管经历了20多年的研究,这一体系中还一直存在着实验不确定、理论预言不一致的问题.
我们用UHV-STM对Ge原子在Si(111)-7×7表面上的初期吸附以及具体吸附位置进行了系统的研究.如图3所示,STM研究结果表明,初期吸附时,单个的Ge原子位于Si(111)-7×7表面的adatom位置,并优先取代有层错半原胞内角位adatom的位置.STM图像中Ge原子所在位置的起伏高度远远小于它的共价半径的大小,证明存在Ge替代角位Si的替代吸附机制.对Ge的各种可能的吸附构型进行理论计算发现,第一性原理计算结果与实验完全符合,解决了之前20多年来Ge在Si(111)-7×7表面上初期吸附位置在理论和实验上一直悬而未决的问题[18].该项工作对Ge/Si半导体体系生长的理解与认识,以及对该体系的结构生长与特性控制具有重要意义.

图2 Si(111)-7×7表面的高分辨STM实验与模拟图像[14] (a),(b)实验中得到的STM图像,对应样品偏压分别为−0.57 V和−1.5 V,隧穿电流分别为0.3 nA和0.41 nA;(c),(d)为Si(111)-7×7的计算模拟STM图,对应样品偏压分别为−0.57 V和−1.5 VFig.2.High-resolution STM images of Si(111)-7×7 surface and corresponding density functional theory simulation[14]:(a),(b)The experimental STM images acquired at(a)−0.57 V and 0.3 nA,and(b)−1.5 V and 0.41 nA,respectively;the brighter triangles are the faulted halves of the 7×7 unit cells;(c),(d)calculated STM images for Si(111)-7×7 surface with sample bias voltage of−0.57 and−1.5 V,respectively.
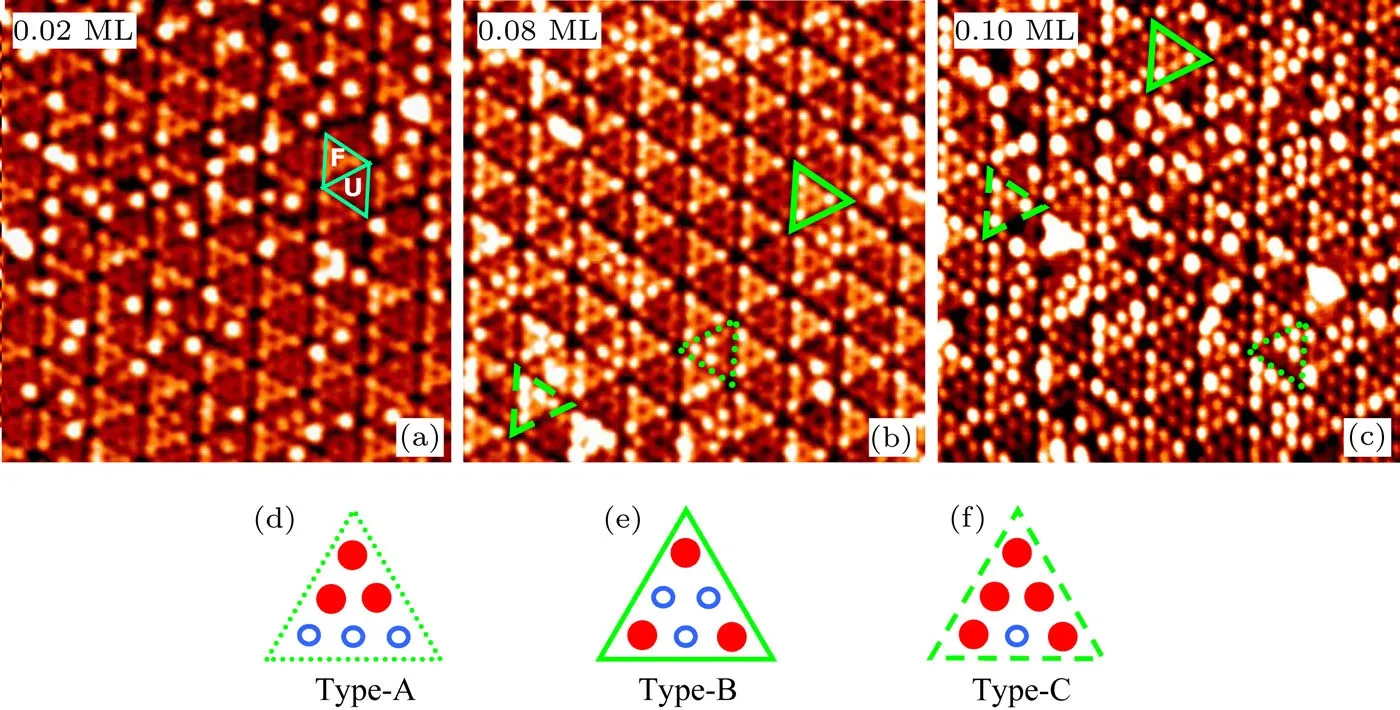
图3 Si(111)-7×7表面Ge原子吸附的STM图像,Ge的覆盖度分别为(a)0.02 ML,(b)0.08 ML,和(c)0.10 ML,图像尺寸为20 nm×20 nm;Ge的三种特征吸附构型分别用点划线、实线和虚线三角形在图(b)和(c)中标出,其对应示意图为(d)—(f)[18]Fig.3.Filled state STM images of the Si(111)-7×7 surface with Ge adsorption.Ge coverages of(a)0.02 ML,(b)0.08 ML,and(c)0.10 ML.A 7×7 unit cell is marked by two triangles in(a),where F and U represent the faulted half unit cell and unfaulted half unit cell,respectively.The scanning areas are 20 nm×20 nm.Three di ff erent con fi gurations of Ge protrusions distributions are denoted in(b)and(c)by the dotted-,solid-,and dashed-triangles,respectively.The schematics for the three typical Ge protrusions,named(d)type A,(e)type B,and(f)type C,are also shown[18].
2.3 固体表面上功能分子的吸附和组装
有机功能分子在光电器件、有机薄膜晶体管和分子器件等方面有广阔的应用前景.分子在基底上的吸附和组装行为是获得低维自组装结构和探索其物性的基础.分子间相互作用和分子与基底间的相互作用在分子组装时相互竞争并最终达到平衡,决定了分子的自组装行为.这两种相互作用对分子在表面上的覆盖度敏感.我们研究了多种有机功能分子在固体表面上的生长及其机制[19−29],本部分着重介绍酞菁铁分子(iron-(II)phthalocyanine,FePc)在Au(111)上从单分子、亚单层到单层和双层的吸附和组装行为.
如图4所示,FePc分子覆盖度很低时,以单分子的形式分散在Au(111)台面上,相对于Au(111)的高对称方向有两种取向.随着覆盖度的增加,FePc分子形成二聚体、三聚体、短链和有独特孔洞的六聚体.在单层时,FePc分子以规则密堆积结构形成了自组装薄膜,薄膜中有相对于Au(111)的高对称方向取向不同的两个畴.双层FePc分子薄膜中FePc分子的形貌与单层薄膜中不同,这是因为与基底相互作用减弱后,分子组装更趋向于分子晶体中的分子组装行为.双层FePc分子薄膜中同样存在着取向不同的两个畴.FePc在Au(111)上的生长行为是由不同覆盖度下分子间相互作用力和分子-基底相互作用力之间的竞争决定的.在低覆盖度下,以分子和基底间的相互作用力为主导,以单分子的形式分散吸附在基底上;在高覆盖度下,以分子间相互作用力为主导,形成规则的自组装结构[26,27].
这一工作可以帮助我们深刻理解FePc分子在Au(111)上的生长行为,为一系列酞菁类分子在Au(111)上的生长提供参考.
2.4 固体表面上功能分子的非模板选择性组装
纳米结构的很多应用是通过有机功能分子的吸附来实现的.可控地、有选择性地在纳米结构的不同表面上吸附具有不同功能的分子,对设计及组装功能纳米结构具有重要的意义.通常,人们采用模板方法来实现纳米结构(包括功能分子纳米结构)的可控制备.如何采用非模板方法实现功能纳米结构的可控自组装和选择性吸附极具挑战性,是纳米电子器件和纳米催化的重要基础问题.
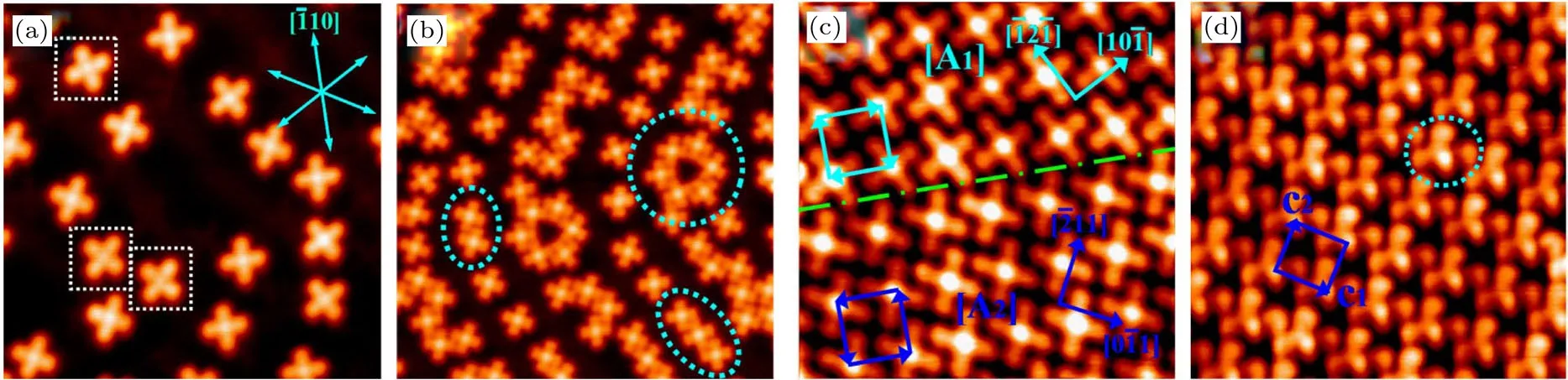
图4 不同覆盖度下FePc分子在Au(111)上的STM图像[26,27](a)0.1 ML FePc/Au(111),FePc分子相对Au(111)有两种取向,虚线方框标出了其中的一种;(b)0.6 ML FePc/Au(111),虚线标出了该覆盖度下特有的分子短链和六聚体;(c)Au(111)上的单层FePc薄膜,相对基底有取向不同的两个畴;(d)Au(111)上的双层FePc薄膜,方框与圆圈分别为薄膜的原胞和一个FePc分子Fig.4.STM images of FePc molecules on Au(111)surface of di ff erent coverages[26,27]:(a)0.1 ML FePc molecules on Au(111)surface,showing dispersed adsorption of FePc molecules with two kinds of adsorption con fi gurations;molecules that are marked with squares and the rest of the molecules belong to di ff erent con fi gurations;14 nm×14 nm;(b)0.6 ML FePc molecules on Au(111)surface,showing initial aggregations that are marked with dashed circles;20 nm×20 nm;(c)the fi rst monolayer of FePc on Au(111),showing ordered molecular arrangements with two di ff erent domains;10 nm×10 nm;(d)the second FePc layer on Au(111),showing an ordered structure;one unit cell and one FePc molecule are marked with a square and a circle,respectively;10 nm×10 nm.
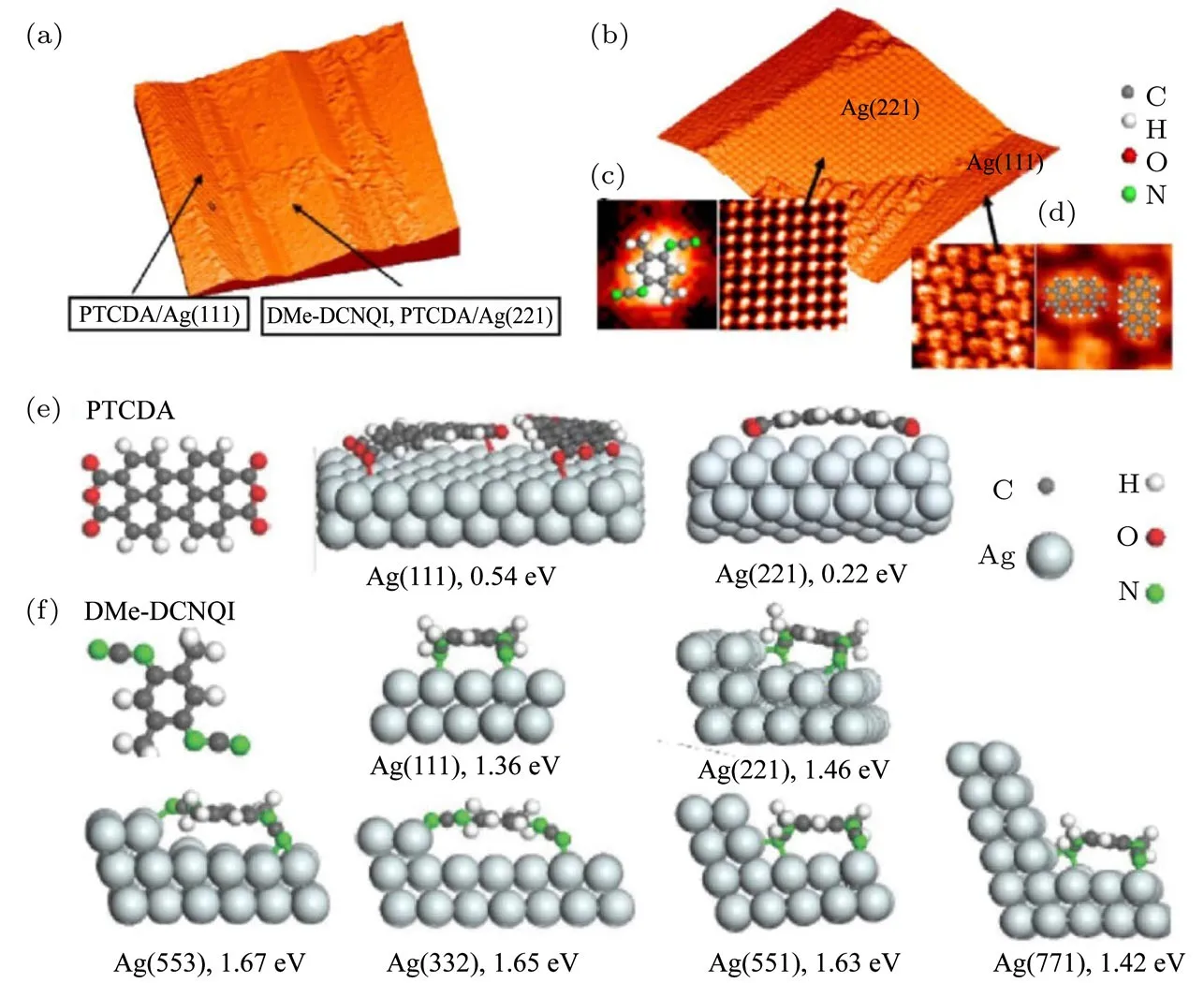
图5 DMe-DCNQI和PTCDA分子在Ag(775)上吸附的STM图像及可能的吸附构型[30] (a)DMe-DCNQI和PTCDA在Ag(775)上的STM图,两种分子共吸附在(221)面上,而(111)面上只有PTCDA分子(50 nm×50 nm);(b)DMe-DCNQI/Ag(221)和PTCDA/Ag(111)的STM图(30 nm×30 nm);(c)DMe-DCNQI/Ag(221)的STM图(右图,6 nm×6 nm)和对应的分子模型(左图);(d)PTCDA/Ag(111)的STM图(左图,6 nm×6 nm)和对应的分子模型(右图);通过密度泛函理论计算优化得到的(e)PTCDA,(f)DMe-DCNQI的分子构型和它们在不同晶面上可能的吸附构型及吸附能Fig.5.The adsorption behaviors of DMe-DCNQI and PTCDA molecules on di ff erent facets of Ag(775)[30]:(a)50 nm×50 nm image of co-adsorbed DMe-DCNQI and PTCDA,showing that both molecules can be found on(221)facets,while(111)facets are only covered with PTCDA molecules;(b)zoom-in image of(a),showing the ordered molecular structures of DMe-DCNQI/Ag(221)and PTCDA/Ag(111),30 nm×30 nm;(c)zoom-in STM image of(b),showing the detailed structure of DMe-DCNQI on Ag(221)(6 nm×6 nm,right panel),and schematic structure of DMe-DCNQI/Ag(221)superimposed on the STM image(left panel);(d)zoom-in STM image of(b),showing the detailed structure of PTCDA on Ag(111)(6 nm×6 nm,left panel),and schematic structure of PTCDA/Ag(111)superimposed on the STM image(right panel);density functional theory calculated optimized con fi gurations of(e)PTCDA and(f)DMe-DCNQI on the Ag(111)surface and on various stepped surfaces.Binding energies are indicated in each panel.
如图5所示,我们从实验和理论上研究了两种有机分子在银单晶不同表面上的吸附行为,发现在Ag(775)基底上,当3,4,9,10-苝四甲酸二酐(perylene-3,4,9,10-tetracarboxylic-dianhydride,PTCDA)分子和2,5-二甲基N,N′-二氰基苯醌二亚胺(2,5-dimethyl-N,N′-dicyanoquinonediimine,DMe-DCNQI)分子先后沉积时,PTCDA分子会吸附在(111)台阶面上,而DMe-DCNQI和PTCDA分子则共吸附在(221)台阶面上;而当分子沉积顺序相反时(先DMe-DCNQI后PTCDA),PTCDA分子会完全吸附在(111)台阶面上,而DMe-DCNQI分子则完全吸附在(221)台阶面上.该研究还阐明了PTCDA分子与基底原子之间的相互作用机制,提出了PTCDA与基底之间的相互作用是通过分子末端官能团-氧原子实现,中心的π共轭区域与基底相排斥,理论模拟得到的氧原子的NIXSW相干长度以及碳原子的K边X射线吸收谱与实验结果符合得很好[30].这一结论否定了Tautz等[31,32]提出的PTCDA与基底间的相互作用是通过其中心的π共轭区域与基底成键的机制.
这项研究开创了一种新的外延生长A/B有序纳米结构的途径——非模板选择性自组装,同时提出了功能基团在选择性吸附中的决定性作用,对二维有序组装和各类纳米功能器件的构造具有重要的指导意义.
本节介绍了纳米尺度“海马”分形结构的形成及其生长机制,提出具有原子尺度大小尖端的针尖可以有效提高STM的实空间分辨率,获得了Si(111)-7×7表面最高分辨的STM图像,并进而解决了Ge原子在Si(111)表面的初期吸附位这一长期悬而未决的问题;阐明了分子在固体表面上的吸附与组装行为,并提出了利用功能基团的不同实现分子的非模板选择性自组装方法.这些工作对表面初期生长、低维有序组装和各类纳米功能器件的构造具有重要价值.
3 纳米量子结构电导和自旋态的可逆转变及其超高密度信息存储
不断完善纳米信息存储材料的功能是当今信息科学的重要研究方向.这类材料需具备更高的存储密度、更快的开关速度以及更好的稳定性和重复性.我们对一系列功能纳米材料进行了探索,致力于设计具有稳定结构与优异特性的有机功能纳米结构,研究其组装结构、有机/无机复合薄膜的纳秒级电导转变、稳定性和重复性以及在超高密度信息存储领域的潜在应用.在此基础上,实现了单个磁性分子在金属表面上的自旋态的量子调控,同时原理性地实现了单分子尺度的超高密度信息存储.
3.1 稳定、重复、可逆的纳米尺度电导转变与超高密度信息存储
对有机功能纳米材料及其纳米级电导相变研究,因其独特的结构和性质可控等优异特性而备受关注.我们在多种有机分子薄膜上实现了基于纳米尺度电导转变的超高密度信息存储[33−39].本小节介绍了一种具有结构双稳态特性的分子——Rotaxane分子.它的特殊结构可以使其在溶液中发生可逆的分子构型改变,从而导致电学特性的转变,即绝缘态和导电态之间的可逆转变.
我们首次报道了在Rotaxane分子固态薄膜中实现分子导电性的转变和超高密度信息存储.通过STM针尖在Rotaxane分子薄膜上施加电压脉冲,在分子尺度上诱导了两个数量级的导电特性转变,在薄膜上重复记录了尺寸为3—4 nm的信息点,如图6所示.研究还发现Rotaxane分子具有非常稳定的电导相变特性.这些结果一方面显示了Rotaxane这种有机功能分子在固体中也同样具有可逆电导转变特性,另一方面为超高密度信息存储研究提供了一类新型的材料[40].
在此工作的基础上,我们进一步改进了原有的Rotaxane分子结构,得到H2 Rotaxane分子.通过在H2 Rotaxane分子薄膜上用STM针尖施加偏压,成功地实现可逆的电导变化和可擦除、稳定、重复的近于单分子尺度的纳米级存储[41],如图7所示.纳米尺度的信息点(∼3 nm)可以在2.0 V,0.1—10 ms的电压脉冲下反复写入,并能在大气室温环境下稳定存在至少12 h.在写入的信息点上施加负电压脉冲(−2.0 V,0.1—10 ms),可以实现信息点的擦除.图7(b)的AFM图像显示,在写入和擦除过程中变化的是H2 Rotaxane分子薄膜的局域电导.
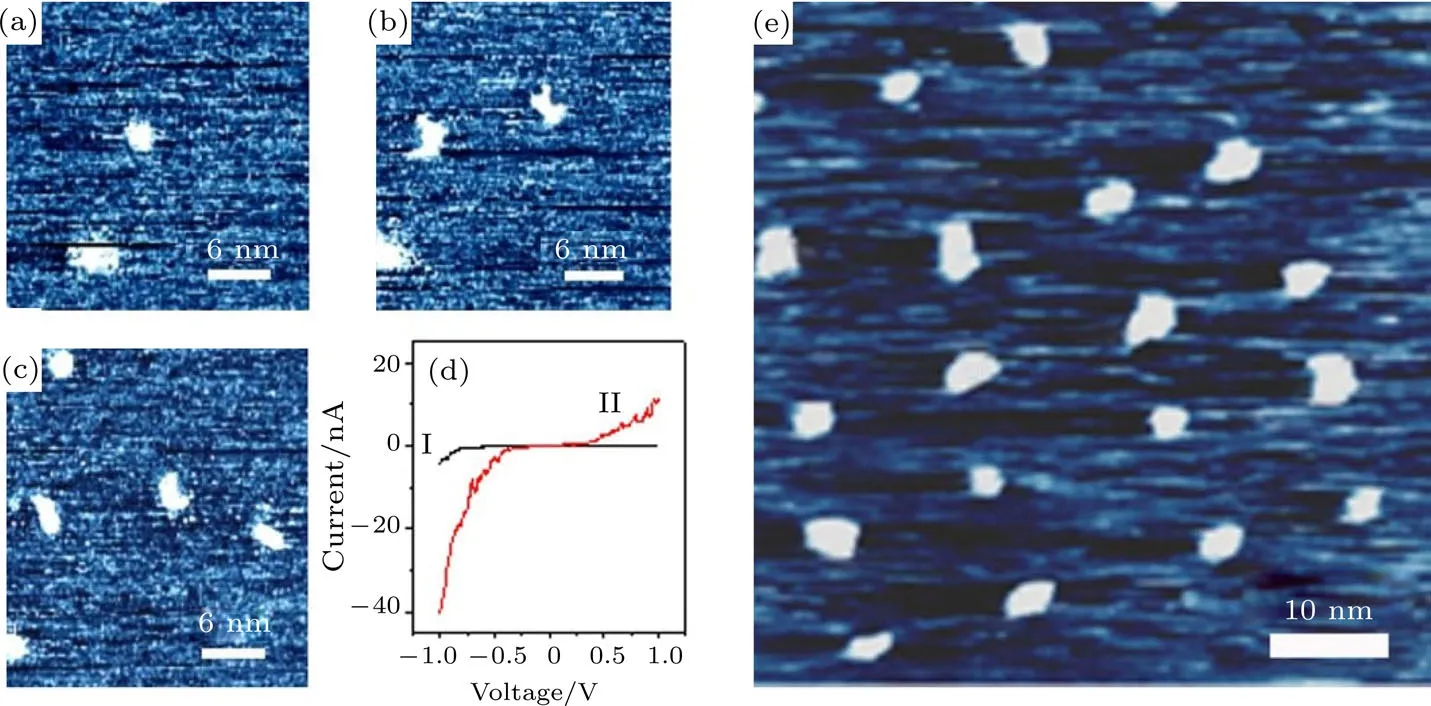
图6 通过STM针尖在Rotaxane分子薄膜上诱导信息点的STM图像[40](a)—(c)通过STM针尖加偏压的方式一个一个地写入信息点;(d)原始薄膜(I)和信息点(II)上的特征I-V曲线;(e)一个5×4的信息点阵,在大气下放置两个月后仍然无损Fig.6.STM images of the recording dots written on the Rotaxane thin fi lms[40]:(a)–(c)Recording dots written one by one through the application of the voltage pulses from the STM tip;(d)typical I-V characteristics measured on the original fi lms(I)and on the induced recording dots(II);(e)STM images of a 5×4 recording dots array on the H1/HOPG sample;this fi lm has been left exposed to air for about 2 months since its preparation.

图7 信息点重复写入与擦除后的STM和AFM图像[41](a)(1)—(3)为用STM针尖顺序写入信息点后的STM图像,(4)—(6)为对信息点的擦除、重新写入和重新擦除后的STM图像;(b)用导电AFM在写入的信息点上得到的形貌像(左边)和电流像(右)Fig.7.STM and AFM images after repeated writing and erasing of the recording dots[41]:(a)Frames(1)–(3)show STM images of bright marks written one by one using STM tip;frames(4)–(6)show STM images after erasing,rewriting,and re-erasing on the same recording site;the voltage pulse for recording was 2 V for 3 ms,the pulse for erasing was−2 V for 3 ms;scale bar is 6 nm;(b)topographic(left)and current(right)AFM images of a mark written by conductive contact AFM on a 8 nm thick H2 fi lms.
为了理解Rotaxene分子在固态薄膜中电导转变是否遵循其在溶液中发生电导转变的机制,即发生分子构型的可逆转变,我们在单个分子和亚分子的水平上研究了单个H2分子的结构与电导转变之间的关系.将H2分子置于Au(111)基底上,直接观察到了Rotaxane分子在外电场诱导下发生分子结构可逆变化同时伴随电导的可逆转变.这是目前为止该类分子结构与电导转变关联的最直接证据,对Rotaxane类分子在分子电子学中的进一步应用具有重要意义[42].
3.2 固体表面上单分子自旋态的量子调控及其原理性应用
近藤效应是指磁性杂质中的局域自旋与自由电子强关联相互作用所引起的电导反常现象.STM技术的迅速发展使人们能够精确地测量单个磁性原子或分子在金属表面上的近藤效应,而在原子尺度上探索影响近藤效应的因素是实现单分子自旋态量子调控的关键.
我们对吸附在Au(111)表面的磁性分子FePc的测量中发现分子中心铁原子在金表面的吸附位置对近藤效应影响很大.如图8所示,FePc在Au(111)表面存在两种吸附取向,虽然在分子中心测量的扫描隧道谱(STS)显示两种分子取向都存在近藤效应,但是彼此却存在很大差别.这种差别主要表现在根据法诺理论拟合的近藤温度,以及STS在费米面附近的线型.第一性原理计算及实验表明,两种取向分子中心的铁原子吸附在金表面的不同位置:第一种取向的分子中的铁原子吸附在金表面两金原子之间的桥位置;第二种取向的分子的铁原子吸附在金表面金原子的正上方.分子中心铁原子在金表面的吸附位置不仅影响到局域自旋与自由电子耦合相互作用的强弱,而且还会影响STS测量中隧穿电子的通道[43].
这是首次报道吸附位置对单分子近藤效应的调控作用.根据该工作观察到的吸附位置对单分子近藤效应的作用,我们提出了调控单分子自旋量子态的可能途径:通过基底上不同位置或不同基底的物理化学性质[44,45]或通过调节纳米分子体系中非功能性侧链[19].这对量子调控和量子信息研究具有重要意义.
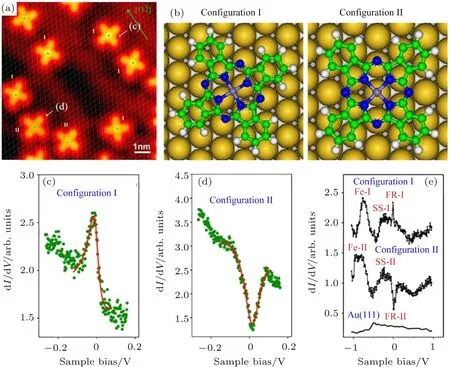
图8 在Au(111)表面两种吸附构型FePc分子的STM图像、构型及Kondo共振谱[43](a)STM图显示Au(111)基底上单分散的FePc分子有两种吸附构型(分别用I和II表示);(b)第一性原理计算优化后的构型I和构型II;(c)FePc分子构型I的dI/dV谱(绿色点线)在费米面附近显示为一个峰,红色实线为法诺拟合的数据;(d)FePc分子构型II的dI/dV谱(绿色点线)在费米面附近显示为一个谷,红色实线为法诺拟合的数据;(e)在FePc分子中心上较大能量范围内的dI/dV谱;Fe-I和 Fe-II是 Fe-3d态;SS-I和SS-II是表面态;FR-I和FR-II是法诺共振Fig.8.STM images and Kondo resonances of two types of FePc molecules on Au(111)[43]:(a)STM image of isolated FePc molecules on Au(111)surface;the overlaid grid represents the gold substrate lattice,showing a shift of 1/2 unit cell between the adsorption sites of the two types(I and II)of molecules;(b)density functional theory optimized molecular con fi gurations of FePc on Au(111);(c)dI/dV spectra(dotted line)for con fi guration I,measured at the center of a type I FePc molecule,showing a peak near the Fermi level,the solid line in the spectra is a fi t of a Fano function;(d)dI/dV spectra(dotted line)for con fi guration II,measured at the center of a type II FePc molecule,showing a dip near the Fermi level;the solid line in the spectra is a fi t of a Fano function;(e)dI/dV spectra scanned with a wider energy range,measured at the center of FePc molecules;Fe-I and Fe-II are the Fe-3 d state;SS-I and SS-II are the surface state;FR-I and FR-II are the Fano resonances.
3.3 具有固定偏心轴的单分子马达及其大面积有序阵列的构筑
我们在金属表面分子器件构筑方面实现了具有固定偏心轴的单分子转子,并实现了其大面积的有序组装[46].以tetra-tert-butyl zinc phthalocyanine((t-Bu)4-ZnPc)在Au(111)表面的吸附为研究对象(图9(a)所示),我们巧妙地利用(t-Bu)4-ZnPc分子与Au(111)表面上的金增原子之间的相互作用实现“抛锚”,通过分子中的N原子和金表面上的Au增原子形成化学键,从而为分子提供了一个固定的转动轴.同时由于分子的四重对称性以及基底的三重对称性,构筑的分子马达每转动30◦会有一个亚稳态,可以模拟马达上齿轮的效果.通过处理Au(111)重构表面使“肘”位(elbow sites)出现增原子,然后沉积(t-Bu)4-ZnPc,就可以实现单分子马达阵列的构筑.图9(b)和图9(c)展示了利用STM观测到的分子马达阵列的形貌像.利用这种方法构筑的单分子马达可以通过控制金增原子的位置、裁剪分子官能团以及替换分子中心金属原子实现转动特性的调控.图9(d)—(f)展示了构筑在Au(111)表面不同重构区域的分子马达,图9(g)和图9(h)分别是(t-Bu)4-ZnPc,ZnPc和FePc在Au(111)形成分子马达的STM图像.图像中不同转动图案以及同一转动中不同的明暗特征,反映了分子马达转动特性的不同.对于利用FePc分子构筑的分子马达,由于中心为磁性Fe原子,马达转动时可以得到在纳米尺度上旋转的磁场,借此可以制作纳米单分子发电机.

图9 单分子马达及大面积有序马达阵列的构筑[46] (a)(t-Bu)4-ZnPc分子吸附在有增原子的Au(111)表面的原子构型;(b),(c)大面积有序单分子马达阵列STM图像;(d)—(f)构筑在Au(111)表面不同重构区域的单分子马达;(g)—(i)利用具有不同官能团的分子构筑的单分子马达Fig.9.Construction of single and arrayed molecular rotors[46]:(a)The adsorption of(t-Bu)4-ZnPc on Au(111)surface with a Au adatom;(b)and(c)STM images of arrayed molecular rotors;(d)–(f)a single molecular rotor at di ff erent regions of a Au(111)reconstruction surface;(g)–(i)a single molecular rotor constructed by molecules with di ff erent functional groups.
小分子机器的构筑和使用过程中会涉及大量的复杂动态过程,准确地识别复杂动态过程中的分子构型是设计和构筑小分子复杂动态量子结构的基础.但由于这个过程通常在室温下发生,系统通常处于多种激发态,且不少动态过程是非原位的,为研究带来了一系列复杂的问题.
我们发展了利用“扫描隧穿电流-时间谱(I-t谱)”与“第一性原理计算”相结合获得小分子复杂动态过程中量子结构的原子构型的方法[20].以FePc分子在Au(111)表面的吸附为例,变温实验证明,由于扩散势垒很小,即使在液氮温度(77 K)下,单个FePc分子也会在Au(111)表面剧烈扩散,这种快速扩散是大尺度和多方向的,导致小偏压下很难获得清晰的STM图像,无法给出体系的原子构型.将针尖固定在有分子经过的区域(图10(a)中箭头所示的蓝点),获得测量隧穿电流与时间的关系(I-t谱,图10(b)),这种I-t谱杂乱无章,传统的I-t谱统计处理方法依赖于统计宽度的选择,具有较大的任意性.我们发展了一种新的排序的I-t谱图(sorted I-t spectroscopy,图10(c))[47,48].这种统计分析可以清楚地给出4种典型的电流值,并且获得分子处于这4种典型电流值的时间比例,进而推断出不同构型之间的能量差,绘制出体系的扩散通道(图10(d)和图10(e)).通过与第一性原理计算得到的稳态与亚稳态之间能量差进行比较(图10(f)),就可以得到这些典型态对应的原子构型.

图10 复杂小分子量子结构动态过程中的构型识别[47] (a)FePc分子在Au(111)表面快速扩散的STM图像,图像尺寸为14 nm×14 nm;(b)在(a)图中蓝色点处获得的I-t谱曲线;(c)针对I-t谱曲线进行的统计分析图;(d)最稳构型与次稳构型能量差及(e)最稳构型与第三稳构型能量差的二维插值图像,色条覆盖50 meV;(f)通过实验I-t谱曲线分析得到的不同构型能量差与第一性原理计算结果的比较Fig.10.Identi fi cation of multiple con fi gurations of small molecule machines[47]:(a)An STM image(14 nm×14 nm)to show the fast di ff usion of FePc molecules on a Au(111)surface;(b)I-t spectrum measured at the site marked with a blue spot in(a);(c)frequency-counting statistical analysis of the I-t spectrum according to current values in panel(b);(d)energy di ff erence between the most stable and second stable con fi guration in each site in a two-dimensional presentation(graph interpolated);(e)energy di ff erence between the most stable and third stable con fi guration in each site in a two-dimensional presentation(graph interpolated);(f)comparison of tunneling current,occupation probability,and energy di ff erence between experiments and density functional theory calculations.
3.4 单个自旋量子态的可逆操控及其在超高密度量子信息存储中的原理性应用
对纳米量子结构的电子态和自旋态的可逆调控及其在未来量子信息中的应用一直是凝聚态物理及其应用领域中最重要的前沿研究之一,相关研究具有极强的挑战性.在3.2节工作的基础上,我们进一步实现了单个自旋量子态的可控且可逆转变,并成功地将其原理性地应用于超高密度的信息存储[49,50].
如图11所示,我们在超高真空STM系统中,通过对Au(111)表面的酞氰锰分子(MnPc)系统通入氢气或解离后的氢气,实现了分子中心磁性原子的单个氢原子吸附,从而实现了该体系近藤效应由“开”到“关”的转变.进一步的密度泛函理论计算表明,虽然氢原子吸附几乎没有改变锰离子的价态,但电子在3d轨道内的重排导致了分子的自旋由S=3/2降为S=1;同时氢原子吸附导致分子与基底的间距增大,使得近藤效应消失.更重要的是,这一自旋态的转变过程是可逆的:通过在局域范围内对STM针尖施加电压脉冲,或者给样品加热,可以实现分子的单个或集体脱氢,从而恢复体系的自旋态和近藤效应[50].我们进一步利用MnPc分子在Au(111)基底上形成的周期性密排阵列实现了单分子尺度(1.5 nm大小的存储点和1.5 nm的最小点间距)的高密度信息存储,为未来在超高密度信息存储与计算等领域的应用开辟了新的途径.
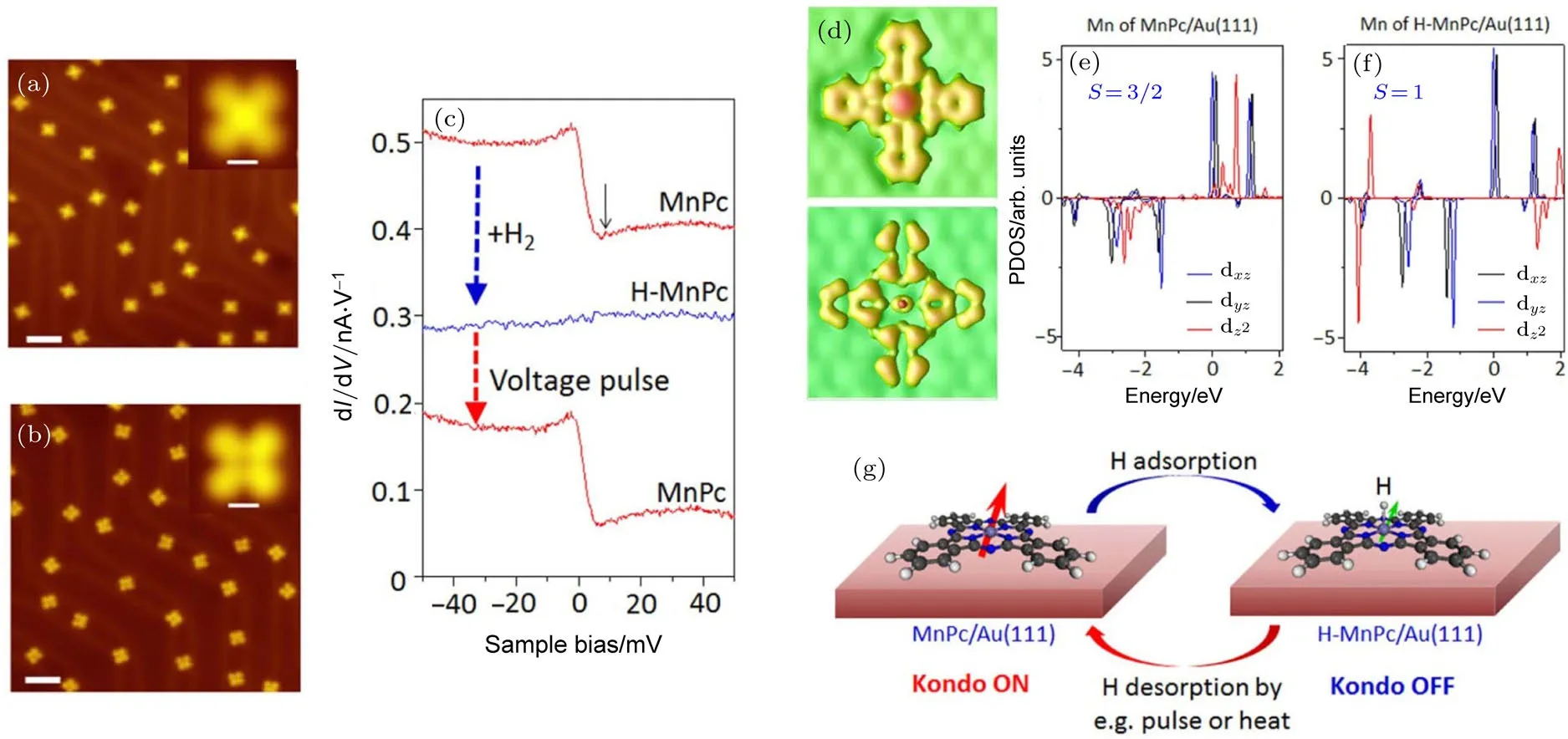
图11 MnPc分子近藤开关[50](a)MnPc分子吸附在Au(111)上,其形貌上中心为亮点;(b)氢原子修饰后,MnPc分子的中心变暗;(c)氢原子修饰前后MnPc分子中心dI/dV谱的变化;(d)MnPc/Au(111)和H-MnPc/Au(111)的STM模拟图,表现出与(a)和(b)中相同的形貌特征;(e),(f)分别为MnPc/Au(111)和H-MnPc/Au(111)中Mn原子的dxz,dyz和dz2轨道的局域电子态密度(PDOS),显示氢修饰后Mn原子的自旋由3/2降为1;(g)通过氢原子的吸附与脱附可以进行可逆近藤开关Fig.11.MnPc Kondo switch[50]:(a)Topography of the MnPc molecules on Au(111)before hydrogen addition,showing molecules with bright center;scale bar,5 nm;(inset)high-resolution image of a single molecule;scale bar,0.7 nm;(b)topography of the MnPc molecules after hydrogen atom decoration,showing some molecules with suppressed center;scale bar,5 nm;(inset)high-resolution image of a single molecule with suppressed center;scale bar,0.7 nm;(c)sequential variation of the dI/dV spectra recorded at the center of a MnPc molecule induced by the absorption and desorption of a single hydrogen atom;for comparison purpose spectra were vertically shifted 0.1 nA/V and 0.33 nA/V for middle and upper curves,respectively;(d)STM simulation of the MnPc/Au(111)and the H-MnPc/Au(111),con fi rming the topographic feature in(a)and(b);(e),(f)PDOS of dxz,dyzand dz2orbitals of Mn ion in the MnPc/Au(111)and the H-MnPc/Au(111),respectively,revealing that spin at the Mn ion is reduced from 3/2 to 1;(g)schematics of reversible control of molecular Kondo e ff ect with adsorption(Kondo OFF)and desorption of hydrogen atom(Kondo ON).
3.5 原子尺度上朗德g因子的空间分辨及其空间分布不均匀性的发现
理解与调控纳米量子结构的自旋特性是自旋电子学领域的前沿研究课题之一.例如,原子的朗德g因子,它反映了其自旋轨道相互作用的信息,可以为分子自旋态的调控提供有价值的参考.对于分子体系,通常的技术手段测得的g因子是大量分子的平均信息,无法得到单个分子的g因子及其空间依赖关系,而对空间依赖关系的研究需要进一步获得单个原子的g因子.因此,如何直接在单原子尺度上测量g因子是描述其空间分布的关键科学问题,也是国际上该领域长期存在的极具挑战性的难题.
一直以来,在对磁性量子体系的研究中,测得的g因子都是在空间上均匀分布的.为了创造g因子在空间分布不均匀的体系,需要设计一种“功能分子”,使其在原子尺度上呈现出可能的g因子的空间不均匀性.在MnPc分子中仅有Mn具有磁性,利用分子“手术”技术对MnPc分子进行剪裁,使得磁性原子Mn周围的C,N等原子也会产生磁性.通过对Mn原子以及C,N原子进行STS的测量,利用近藤效应(Kondo e ff ect)随磁场变化的实验可以获得不同磁性原子(Mn,C或N)的g因子,从而在单原子尺度上实现g因子的空间分辨.原子处在晶体场(crystal fi eld)中时,自旋轨道耦合作用会将激发态的轨道角动量本征态与基态混杂,从而使原子的g因子偏离处于自由状态时的值.因此,通过单分子内部不同位置处的g因子的大小,就可以得到自旋轨道耦合作用在单分子尺度的空间分布信息[51].
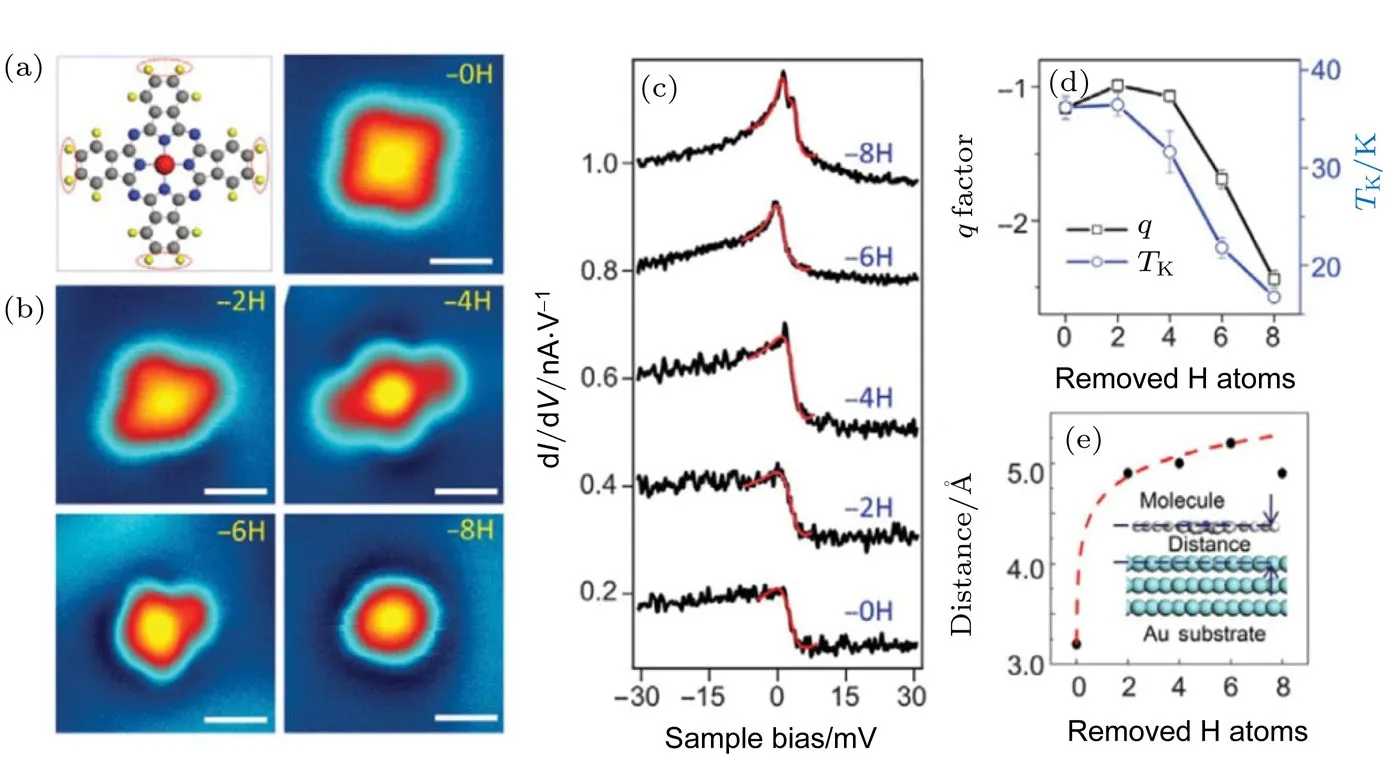
图12 逐步脱氢后的STM,STS,q因子,TK以及分子与基底间距随脱氢数量的变化趋势[51](a)分子模型(左)和完整的MnPc分子的STM图;(b)逐步脱氢后MnPc分子的STM图,分子外缘的四对氢原子(如图(a)中红色点线所示)在STM针尖脉冲的作用下逐步脱去;(c)图(a)和(b)中对应分子的dI/dV谱,所有的谱线均在中心Mn原子上获得;红色曲线为法诺拟合的近藤共振曲线;(d)通过法诺拟合的dI/dV谱得到的近藤温度和q因子随着脱氢数量的变化;(e)计算得到的中心Mn原子和Au基底的距离随脱氢数量的变化,红色虚线为指数拟合Fig.12.The STM images,STS,q factor and TKduring systematic dehydrogenation and the relationship between molecule-substrate distance and hydrogen atom numbers[51]:(a)Molecular model(left)and STM images of intact MnPc molecules;scale bar,1 nm;(b)STM images of dehydrogenated MnPc molecules;the pairs of outer hydrogen atoms of each of the four lobes(as indicated by dotted circles in(a))were removed step by step;scale bar,1 nm;(c)corresponding dI/dV spectra of molecules in(a)and(b);all spectra were acquired at the position of central Mn ions;each successive plot is vertically shifted for clarity;the red curves are Fano fi ttings of the Kondo resonance as described in the main text;(d)evolution of Kondo temperature(TK)and q factor with the number of removed H atoms,derived from Fano fi tting of dI/dV spectra;(e)calculated distance between central Mn ion and Au substrate(the fi rst layer)with the increase of number of H atoms removed;the red dashed line shows an exponential fi t.
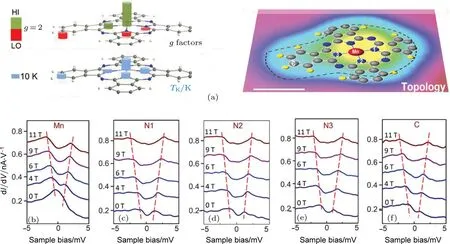
图13 朗德g因子原子尺度的空间分布[51](a)—6H-MnPc分子上朗德g因子分布、近藤效应转变温度分布、以及STM形貌像(从左到右),标尺为0.5 nm;(b)—(f)分子内部不同原子处的STS谱对外加磁场的响应Fig.13.Site-dependent g factor of a—6H-MnPc molecule[51]:(a)Three-dimensional stacking map showing topographic image,discrete g factor,and TKin di ff erent two-dimensional layers;topography of a—6H-MnPc molecule is overlaid with schematic molecular model;scale bar,0.5 nm;(b)–(f)dI/dV spectra acquired at di ff erent atomic sites speci fi ed in(a);all dI/dV spectra were measured at the sample temperature of 0.4 K under a magnetic fi eld of Bz=0–11 T.
如图12所示,我们首先利用STM针尖对吸附在Au(111)表面的MnPc分子施加电压脉冲,进行逐步脱氢.去掉氢原子的苯环与金基底成强相互作用使得构型发生弯曲,与分子骨架相比距离更近.脱氢后的MnPc分子展现出了扩展的近藤效应,即在分子的非磁性原子部分也可以探测到Kondo共振.密度泛函理论计算表明,脱氢后分子骨架对称性降低,从而导致了自旋极化的出现,自旋极化作用使得部分C,N原子产生磁性.通过测量脱氢后分子不同位置的Kondo共振峰在磁场下的劈裂,就得到了g因子的空间分布(图13).实验上发现,g因子的分布是不均匀的,表明了不同量子数的原子轨道的自旋轨道耦合作用是不同的[51].这项工作为探索单分子内部的自旋相关特性提供了新的途径.
本节介绍了实现超高密度信息存储的两种思路:利用有机分子薄膜中纳米尺度电导转变和通过单个磁性原子或分子自旋态的转变实现超高密度信息存储.前者可以通过设计具有优良结构与特性的有机分子来实现,后者可以通过控制分子在表面的吸附构型、修饰分子中心的磁性原子或者改变分子构型来达到目的.在此基础上,我们还通过分子“手术”的方法研究了朗德g因子在原子尺度上的空间分辨,发现其空间分布具有不均匀性.这些工作为未来有机功能分子在超高密度信息存储和量子调控等方面的应用奠定了基础.
4 石墨烯及几种新型二维原子晶体材料的可控构筑和物性调控
石墨烯是一种由碳原子构成的蜂窝状单层结构.2004年,Geim和Novoselov用剥离方法成功制备了石墨烯并发现了其新奇的量子特性[52],2010年他们因此获得了诺贝尔物理学奖.石墨烯具有超高的载流子迁移率、超高的透光率、室温下的量子霍尔效应等优良特性,使其在电子学、光学、磁学、催化、储能和传感器等领域有着广阔的应用前景,被称为“未来材料”、“变革性材料”[4,52].石墨烯的成功引发了世界范围内对新型二维晶体材料的探索和研究热潮.
我们在石墨烯及类石墨烯二维原子晶体材料的制备、物性与应用基础等方面开展研究,取得了一系列国际前沿性研究成果.在过去的十年间,我们采用分子束外延生长方法制备出了晶圆尺寸、高质量、单晶石墨烯[53−56]及类石墨烯二维原子晶体
材料[57],包括: 硅烯[58,59]、锗烯[60,61]、铪烯[762]、二硒化铂[63]、锑烯[64],HfTe3/HfTe5异质结[65]和图案化的二硒化铂与硒化铜[66]等.本节将详细介绍这些工作.
4.1 晶圆尺寸、高质量、单晶石墨烯的制备及原位硅插层绝缘化
石墨烯的广泛研究得益于其单层样品的成功制备.过去几年中典型的制备方法主要有机械剥离天然石墨[52],SiC单晶外延[67]以及化学气相沉积(CVD)[68]法.基于机械剥离方法制成的样品,由于没有外延基底的影响,能够反映石墨烯的本征物理性质.但是,这种方法效率低,加上天然石墨本身不是完美的单晶结构,其晶粒尺寸通常在几个微米以下,导致其不能应用于石墨烯样品的大规模制备.高温分解SiC单晶法外延石墨烯是另一种重要方法,这种方法效率较高,但样品存在大量的缺陷.CVD法制备石墨烯方法简单,适合大规模制备,但是得到的石墨烯缺陷多,电学、力学性能都较差.因此,获得高质量、大尺寸的单晶石墨烯材料,对于石墨烯本征物理特性的基础研究及其在未来器件中的应用具有极强的挑战性和重要的科学意义.
在高质量单晶石墨烯薄膜的生长制备方面,我们发现含碳的Ru(0001)单晶在超高真空环境下经高温退火处理可以使碳元素向晶体表面偏析形成外延单层石墨烯薄膜.初步结果于2007年报道[53],得到国际同行的广泛关注,是国际上最早使用该方法制备石墨烯薄膜的两个课题组之一[69].此后,我们通过优化生长条件获得了高质量、毫米级尺寸的外延石墨烯单晶,并通过一系列实验与理论研究证实了其大面积、高质量与连续性[54].如图14所示,在厘米尺寸单晶上不同位置的低能电子衍射(low-energy electron di ff raction,LEED)结果证实了石墨烯样品的毫米级的高度有序性;STM的研究结果表明石墨烯在基底表面形成六角排布摩尔超结构,高分辨的STM图显示了这种超结构原胞内每个原子的位置,并且证实了石墨烯薄膜沿基底台阶的连续性生长.这种高质量的石墨烯单晶结构的获得是我们在该研究领域中独具特色的工作,为石墨烯基础问题的深入研究及其进一步在器件方面的应用提供了一种新方法和理想体系.
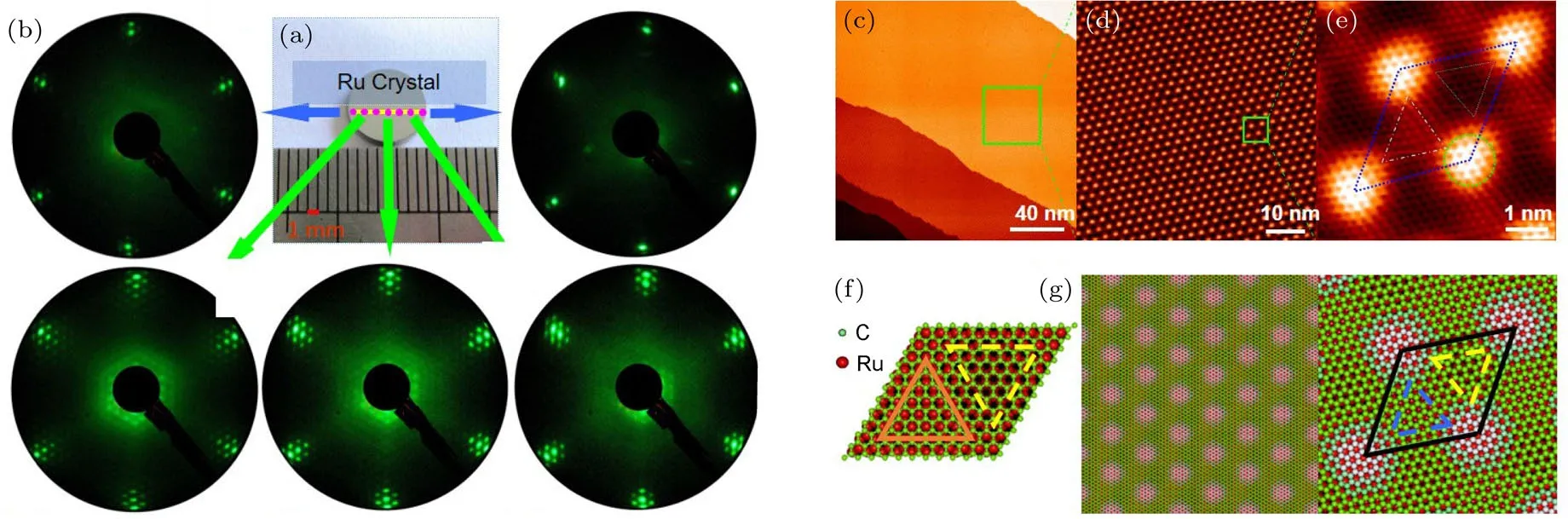
图14 Ru(0001)单晶表面外延生长大面积、高质量、单晶石墨烯的LEED图、STM图及原子结构图[54] (a)和(b)为在graphene/Ru(0001)样品表面不同位置的LEED图样,(a)样品是一个直径8 mm的圆片,电子束沿图中黄色直线移动,粉色点为(b)中各LEED图样对应的位置;(b)中上排的两幅LEED图样在样品的边缘取得,石墨烯的衍射点很弱,下排三幅LEED图样中石墨烯的衍射点强而锐利,这说明除边缘外样品表面覆盖了满层石墨烯;(c)—(e)为样品的STM图,分别为大面积图、石墨烯摩尔点和石墨烯原子分辨,(e)中的菱形为石墨烯摩尔点的一个原胞;(f)和(g)为优化后的原子模型Fig.14.LEED pattern,STM image and atomic structure of large-scale,high quality and single-crystalline graphene epitaxially grown on Ru(0001)[54].(a)and(b)are LEED patterns obtained from di ff erent locations on the sample.(a)The sample is a disk of diameter 8 mm,as shown in the photograph.The electron beam was moved along the yellow line to obtain the di ff erent LEED patterns.The pink spots are the locations where the photographs shown in(b)were taken.In the two LEED patterns in the upper row,the additional spots are weak,but in that in the lower row,the additional spots are sharp,and have the same arrangement.This indicates that graphene formed at all locations,except at the edges.(c)STM image shows the atomically fl at graphene fl ake extended over entire Ru terraces.(d)The hexagonal Moiré pattern formed by the superposition of graphene and the Ru substrate.(e)Atomic-resolution image of one unit cell of the Moiré pattern.(f)Schematic illustration of the optimized atomic structure,showing one unit cell of the superlattice.(g)An enlarged perspective view of the superlattice.
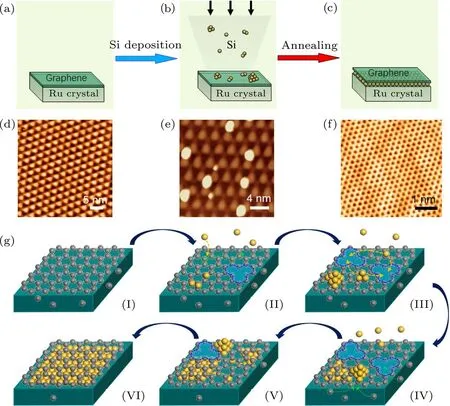
图15 Ru(0001)单晶表面外延石墨烯的硅插层示意图、STM图像和插层机制示意图[70,73] (a)—(c)硅原子在石墨烯/钌界面插层的示意图,包括(a)大面积高质量石墨烯的生长,(b)硅原子沉积在石墨烯表面,以及(c)退火后硅原子插层到石墨烯/钌的界面;(d)—(f)是与(a)—(c)过程相对应的STM图像;(g)硅原子协同作用插层机制的示意图Fig.15.Schematic,STM images and mechanism of Si intercalation between graphene and Ru(0001)[70,73]:(a)–(c)Schematic of the intercalation process including(a)formation of large-scale,high-quality graphene on Ru(0001),(b)deposition of Si atoms on graphene and(c)intercalation of Si atoms between graphene and Ru(0001);(d)–(f)corresponding STM images of the intercalation procedure with respect to(a)–(c);(g)schematic of the cooperative intercalation mechanism.
在这种高质量单晶石墨烯的基础上,我们发展了石墨烯的原位硅插层绝缘化方法[70−72].将半导体硅材料沉积在石墨烯表面,通过高温处理,把硅材料插入到石墨烯与金属之间,形成石墨烯/半导体硅/金属的异质结构,如图15所示.这种方法可以有效避免化学转移方法引入的缺陷和杂质,保证石墨烯原始状态的高质量.利用硅插层技术,可以得到不含电荷杂质和原子级平整的石墨烯硅插层结构,避免了由于结构起伏和电荷杂质对石墨烯载流子的散射,使石墨烯更接近本征的状态.进一步的拉曼和角分辨光电子能谱实验证明,经过硅插层后的石墨烯仍然是高质量、大面积的石墨烯.这种石墨烯原位硅插层绝缘化方法可以把石墨烯材料与现有的硅技术相融合,为石墨烯新型器件研究开辟了一条新途径.
我们进一步研究了石墨烯硅插层的插层机制,提出了插层过程的“协同作用插层机制”[73].该机制包括以下5个关键步骤(图15(g)):1)在硅原子和钌基底的协同作用下,原本无缺陷的石墨烯表面打开碳原子空位缺陷(过程II);2)在空位缺陷和钌基底的协同作用下,硅原子扩散到石墨烯/钌的界面,实现插层(过程III);3)插层后的硅原子在界面扩散形成规则的二维结构(过程IV);4)钌基底中的碳原子偏析到表面修复石墨烯中的空位缺陷,使石墨烯恢复原始状态的高质量(过程V);5)随着插层硅原子的增加在石墨烯/钌界面形成单层硅插层结构(过程VI).我们利用STM表征结合第一性原理计算证实了该插层机制的合理性,并进一步将这种插层机制扩展到包含其他基底和插层原子的石墨烯插层体系中,证实了该插层机制具有普适性.
此外,我们还在Pt(111)[55],Ni(111)[74],Ir(111)[56]等多种金属单晶基底上成功地制备出石墨烯并进行了结构和性能的研究.利用这些石墨烯材料,进行了石墨烯上的金属插层[75−79]、分子吸附与自组装[80−85]、掺杂[86,87]和大面积双层石墨烯的构筑[88]等一系列探索,为石墨烯材料的功能化和应用奠定了基础.
4.2 硅烯和锗烯的可控构筑、生长机制与物性调控
硅烯和锗烯具有和石墨烯类似的蜂窝状结构,作为新型二维原子晶体材料,引起了越来越多人的关注和研究.与石墨烯不同,硅烯和锗烯的两个子晶格上的原子在垂直于材料所在平面的方向上有位移,因而形成低翘曲(low buckled)的结构.硅烯和锗烯的可控制备近年来一直是二维材料的研究热点之一.
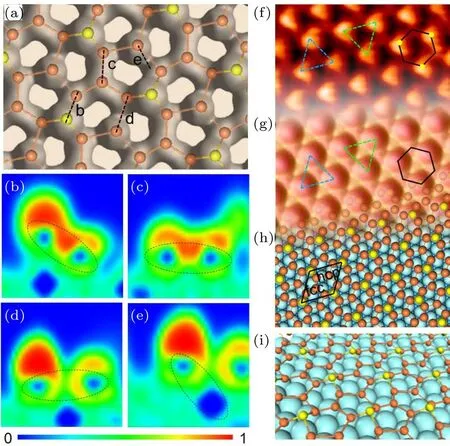
图16 硅烯的原子结构、电子局域函数及STM图像[58](a)硅烯的电子局域化函数(ELF),证明生长出的硅烯是一层连续的薄膜;(b)—(d)沿(a)中四条黑色虚线剖面的ELF,其中(b)为顶位与桥位的硅原子,(c)为fcc与桥位的硅原子,(d)为hcp和桥位的硅原子,它们都表现出共价键连接的特性;(e)硅原子和基底原子间的ELF,表现为静电力相互作用;(f)硅烯薄膜的STM图像;(g)对应的STM模拟图,与实验得到的STM图像完全符合;(h)siliceneIr(111)的原子结构图;(i)为(h)对应的硅烯在Ir(111)表面的图像Fig.16. Atomic structure,electron localization function(ELF),and STM image of silicene on Ir(111)[58]:(a)Top view of the overall ELF of the relaxed model with an ELF value of 0.6,demonstrating a continuity of the silicene layer;(b)–(d)the ELFs of the cross section between silicon pairs,showing the covalent interaction existing between each pair of silicon atoms;the pairs are depicted by the dashed-line ellipses;(e)ELF of the cross section between the silicon atom at hcp hollow site and its nearest iridium atom;the ELF value here is at the range of green-blue region(about 0.38),indicating an electrostatic interaction;the color scale is shown at the bottom;(f)zoomed-in STM image of the silicon layer;besides the brightest protrusions,two other regions showing di ff erent contrast are indicated by the upward and downward triangles;the honeycomb feature is indicated by the black hexagon;(g)simulated STM image,showing features identical with the experimental results in the same triangles and hexagons;(h)top view of the relaxed atomic model of the siliceneIr(111)con fi guration;(i)perspective view of the relaxed model in(h),showing an undulated silicene on Ir(111)surface.
由于石墨层间存在弱的范德瓦耳斯力,可以很容易地从石墨中解理出石墨烯;但硅烯和锗烯则不能从体相的硅和锗中获得,这是因为在体材料中,硅(锗)原子间是以sp3杂化轨道形成的共价键相连,这种原子间强的共价作用很难被破坏,通过解理获得硅烯和锗烯是不太可能实现的.因此,在固体表面外延生长成为了一种主要的制备方法.
对于硅烯的研究主要集中于在Ag(111)基底上的生长.然而,在Ag(111)基底上制备出来的硅烯有多种畴,畴与畴之间的晶界无疑会影响到硅烯的本征性质,这也是目前基于硅烯的场效应晶体管的电子迁移率远远低于理论预期值的原因之一.因此,研究硅烯的生长机制、探索合适的基底生长大面积、高质量的硅烯对于基础研究和应用探索都具有十分重要的意义.
我们通过外延的方法在金属基底Ir(111)表面成功地制备出了硅烯.通过LEED和STM的表征,发现它相对于金属铱基底表现为一种的超结构.这种结构恰好是和硅烯的超结构相匹配的.第一性原理计算验证了这种超结构模型并提出这是一层起伏的硅烯.重要的是,计算得到的电子局域函数显示,相邻硅原子间的局域电子显著多于硅原子与基底的局域电子,从而证明Ir(111)表面外延生长的硅烯是一层连续的二维原子晶体(图16),而非简单的硅原子吸附结构[58].
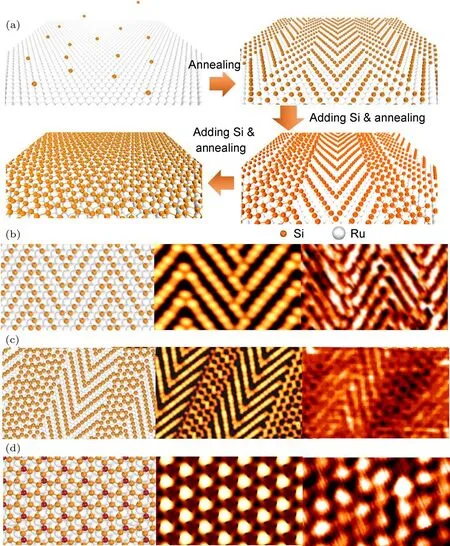
图17 Ru(0001)基底上硅烯的生长机制[59] (a)实验过程示意图;(b)—(d)分别为硅鱼骨结构、鱼骨-硅烯纳米带超结构和硅烯的优化后的原子结构图、STM模拟图和实验中的STM图Fig.17.Growth mechanism of silicene monolayer on Ru(0001)[59]:(a)A schematic to show the formation of Si monolayer structures formed on Ru(0001);(b)–(d)relaxed atomic model,STM simulated image and STM image(from left to right)of the silicon herringbone structure(b),silicon herringbone-hexagonal superstructure(c),and silicene(d).
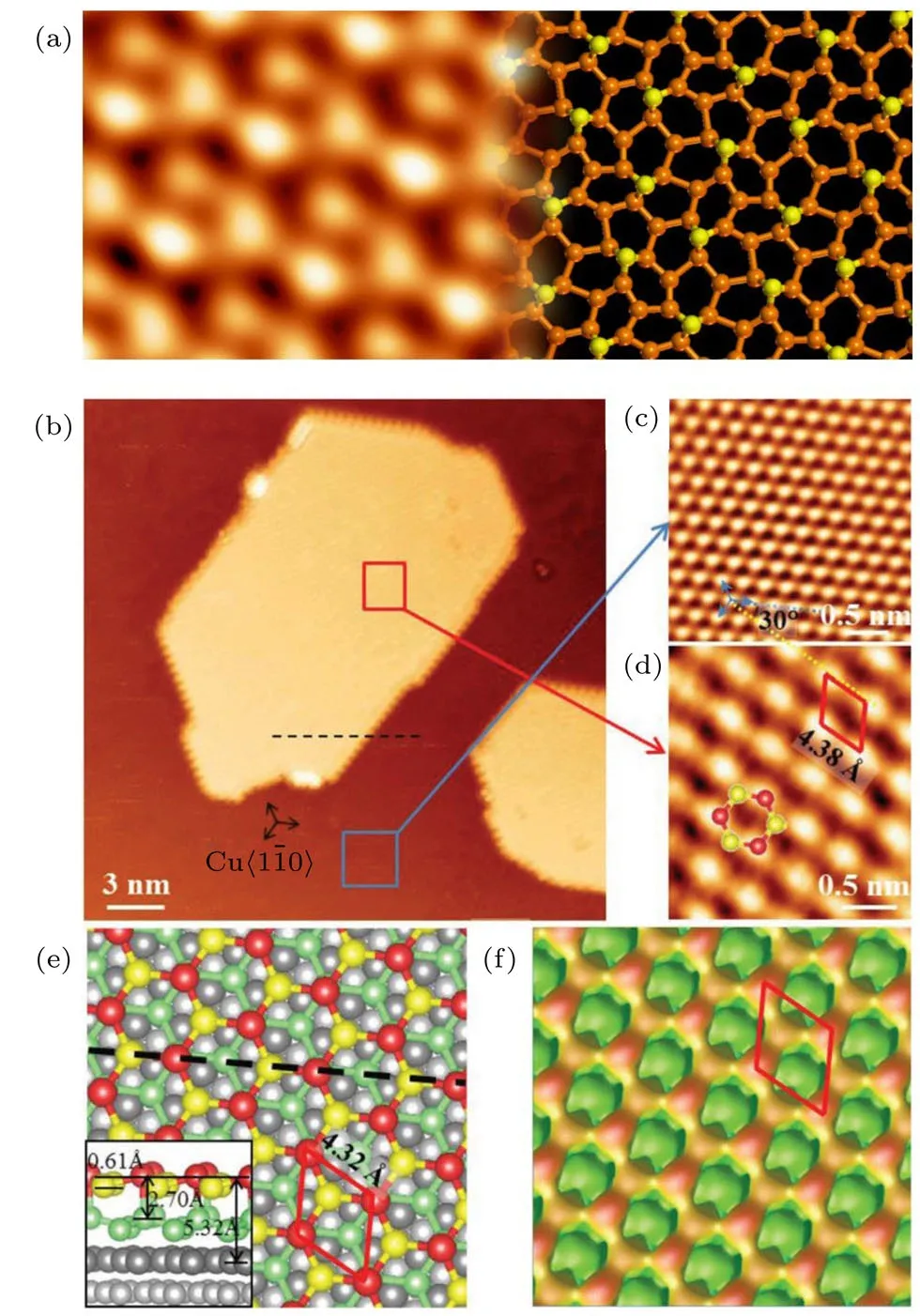
图18 单层和双层锗烯的STM图像、结构图及STM模拟图[60,61] (a)Pt(111)上锗烯的STM 图(左)和优化后的结构图(右);(b)—(f)Cu(111)上双层锗烯的STM图、结构图和STM模拟图,其中(b)为大尺度双层锗烯的STM图;(c)为原子分辨的Cu(111)基底STM图,锗烯岛边界的取向与基底的高对称方向Cu⟨1¯10⟩平行;(d)为具有原子分辨的双层锗烯的STM图,锗烯与基底形成周期为4.38 Å的超结构,黄色与蓝色点划线之间的夹角为30◦;(e)优化后的双层锗烯在Cu(111)上的结构顶视图,内嵌的侧视图显示,上层锗烯的起伏为0.61 Å,两层锗烯的间距为2.7 Å;(f)为STM模拟图,图像与实验一致,表明所制备样品确实为双层锗烯Fig.18.STM image,atomic model and STM simulated image of mono-and bi-layer germanene[60,61]:(a)Germanene on Pt(111);(b)–(f)biatomic-layer germanene on Cu(111);(b)unoccupied-states STM image,showing well-shaped 2D germanium nanosheets;the perimeters of the germanene sheets run parallel to the close-packed direction Cu⟨1¯10⟩,deduced from(c)the atomic resolution STM image of the pristine Cu(111)(blue contour);(d)zoomed-in STM image of a germanium adlayer(red contour),revealing the periodicity of the germanene superstructure(4.38 Å);the angle between the blue and yellow dotted lines is about 30◦;(e)schematic illustrating(top view)the relaxed atomic model of theR30◦BL germanene/Cu(111)con fi guration;(inset)side view of the con fi guration reveals a corrugation of around 0.61 Å in the adlayer and the interlayer distance between the two germanium layers of 2.70 Å;one unit cell is indicated as a red rhombus;(f)simulated STM image with the features fi tting very well with the experimental observations.
除Ir(111)基底外,还发现在Ru(0001)基底上可以制备出硅烯,并且系统地研究了硅烯在Ru(0001)基底上的生长及其机制.如图17所示,在Ru(0001)基底上,硅原子首先沉积在基底的hcp hollow位置,形成线性结构.因为受到具有三重对称性的基底调制,硅原子并不会形成无限长的链状结构,而是形成一种之前从未被发现过的硅鱼骨结构.随着硅原子沉积量的增加,硅增原子会优先吸附于硅鱼骨的elbow位置,在elbow位置形成硅的六元环.继续增加硅原子的沉积量,这些六元环会作为成核点在硅鱼骨结构的elbow区域转变成小片的硅蜂窝状结构(硅烯纳米带),最初形成的鱼骨结构演变成鱼骨和蜂窝状结构共存的二维超结构.进一步增加硅原子的沉积量,硅鱼骨结构消失,整个基底被硅蜂窝状结构——即硅烯——所覆盖[59].
另外,我们还首次在Pt(111)基底上制备出锗烯[60],随后在Cu(111)基底上制备出双层锗烯,并开展了结构与电子结构特性研究[61],如图18所示.高分辨STM图像结合第一性原理计算证实,Pt(111)上的锗烯为低翘曲结构,相对于基底形成的超结构.Cu(111)上的双层锗烯为AB堆垛,与基底形成的超结构.由于底层锗烯有效屏蔽掉了来自铜基底的相互作用,双层锗烯在费米能级附近呈现完美对称的“V”形dI/dV电子态,表现出自由锗烯才有的线性能带色散特征.
这些工作提供了制备高质量硅烯和锗烯的方法,为在硅烯和锗烯中观察到新奇的量子现象提供了可能.但是,由于硅烯和锗烯与现有衬底有较强的相互作用,寻找弱相互作用衬底生长硅烯和锗烯仍然是我们未来工作的重点.
4.3 铪烯的可控构筑
从现有报道的单层二维蜂窝状材料来看,它们只是由元素周期表中的p区元素(非金属元素)所构成的(例如碳元素构成的石墨烯,硅元素构成的硅烯等).含有d电子的过渡金属元素要远多于p区元素.过渡金属元素具有丰富的多体物理和配位化学性质,而且部分元素还含有自旋极化的磁性特性,其二维蜂窝状结构的实现对于研究过渡元素电子、自旋和催化性质等具有重要意义.然而,由d电子过渡金属元素单质构成的二维蜂窝状结构至今未见报道.
过渡金属元素中的铪也是当今半导体科学和技术中最重要的元素之一,我们通过分子束外延生长的方法把铪沉积在Ir(111)基底表面.如图19所示,铪原子在Ir(111)基底上形成了二维蜂窝状晶格,并且相邻铪原子的间距与体相铪内部的原子间距相近.这些结果突破了二维蜂窝状结构主要由p区元素(例如碳、硅)构成的现状.我们对实验结果进行了理论计算与模拟,结果揭示了最近邻铪原子之间是共价键结合.这类由元素周期表中d区元素构成的二维晶体材料,其几何结构和成键方式与石墨烯类似,被称之为d电子烯或者金属烯-铪烯[62].
该工作向实现非碳元素的类石墨烯二维蜂窝状结构迈出了重要的一步.这种d电子金属元素构成的二维蜂窝状结构比石墨烯具有更强的自旋轨道耦合,为研究二维体系中新的量子现象和电子行为提供了全新的平台.
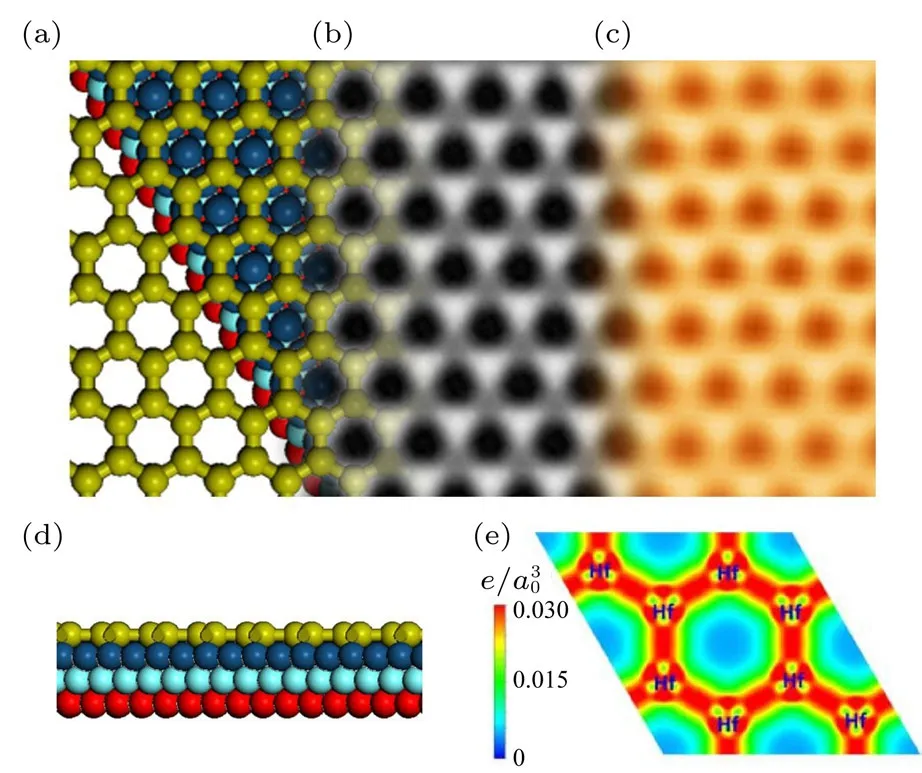
图19 单层铪烯的原子结构与STM图[62](a)—(c)分别为铪烯的优化后的原子结构图、STM模拟图和实验中的STM图;(d)为铪烯原子结构侧视图;(e)为铪烯的二维电荷密度图Fig.19. Atomic con fi guration and STM image of hafnene lattice on Ir(111)[62]:(a)Top view of the calculated atomic structure;half of the Hf atoms are on the fcc sites(vertically above the red Ir balls),while the other half are on the hcp sites(vertically above the cyan Ir balls);for clarity,some of the substrate atoms are removed;the white rhombus denotes the Ir(111)-(2×2)unit cell;(b),(c)density functional theory-simulated and experimental-resolved STM images,showing the honeycomb structure of hafnene lattice;(d)side view of the relaxed atomic structure in(a),showing a planar Hf layer on an Ir(111)surface;(e)the two-dimensional charge density in the Hf plane on Ir(111)substrate;Hf—Hf bonds are clearly seen and they are responsible for the observed honeycomb structure.
4.4 锑烯的可控构筑与稳定性测试
由元素周期表中V族元素构成的层状材料,例如黑磷,具有较大的带隙以及高的载流子迁移率等特性.然而,黑磷在大气条件下不稳定,用它制备的器件的适用性受到限制.最近,V族元素锑构成的单层锑烯(antimonene)引起了研究者的极大兴趣.已有理论预测锑烯的带隙随层厚的改变而变化,特别对于单层锑烯,理论预言其带隙为2.28 eV.同时,与石墨烯相比,锑烯具有更高的载流子迁移率.基于这些特性,锑烯在相关电子器件和光电子器件领域有着潜在的应用前景.因此,如何制备高质量的单层锑烯备受关注.

图20 单层锑烯的原子结构、电子结构与STM图[64] (a)—(c)分别为锑烯的优化后的原子结构图、STM模拟图和实验中的STM图;(d)锑烯原子结构侧视图;(e)锑烯的电子局域化函数(ELF)分布图;(f)沿(e)中黑色虚线的剖面ELF图Fig.20.Density functional theory calculations and atomic con fi guration of monolayer antimonene on a PdTe2substrate[64]:(a)Top view of the relaxed model that has the lowest binding energy of the antimonene with the PdTe2substrate;orange,gray,and cyan balls represent Sb,Te,and Pd atoms,respectively;for clarity,some of the substrate atoms are omitted;(b)simulated STM image,showing features remarkably consistent with the experimental results in the same triangular sublattice;the dashed line rhombus denotes two triangular sublattices of one unit cell in the antimonene;(c)atomic resolution STM image of the antimonene layer;(d)side view of the relaxed structure of antimonene on PdTe2;the distance between the top and bottom Sb sublayers is 1.65 Å,and the distance between the bottom Sb plane and the top Te plane of PdTe2is 2.49 Å;(e)top view of the overall ELF of the relaxed model with an ELF value of 0.6,showing the continuity of the monolayer antimonene;(f)ELF of the cross section along the black dotted line in(e),demonstrating high localization of the electrons in Sb-Sb pairs.
考虑到层状的过渡金属二硫属族化合物(TMD)PdTe2表面化学性质稳定,同时具有六方对称性,晶格周期(4.10 Å)与锑单晶层内的周期(4.12 Å)晶格匹配,与理论预言的单层锑烯(周期4.01 Å)的晶格失配度也只有2.3%.因此,以PdTe2作为基底,利用分子束外延生长方法,我们成功获得了高质量单层锑烯,如图20所示.借助LEED和STM等手段对所生长的单层锑烯的精细原子结构进行了研究,从STM图可以清晰地分辨出锑原子形成了六角蜂窝状结构;LEED实验证明我们获得了大面积、高质量的锑烯单晶.X射线光电子能谱实验和电子局域函数理论计算,揭示了单层锑烯和基底之间为弱的范德瓦耳斯相互作用.进一步的STM和XPS实验观测结果表明,单层锑烯在空气中具有高的化学稳定性,暴露空气后没有被氧化,这一特性对于锑烯进一步走向实际应用至关重要[64].
这一工作提供了一种制备高品质单层锑烯的方法,也提供了一种制备具有原子级平整界面的基于二维材料的异质结构的新思路,即直接利用TMD材料作为基底外延生长单层二维原子晶体材料,为二维材料异质结器件的研究提供了有价值的参考.同时,锑烯作为类石墨烯结构的新型二维原子晶体材料,拓展了非碳基二维蜂窝状原子晶体材料的研究领域,而且它具有宽带隙、高迁移率的特点,在大气环境下也能够稳定存在,因此,锑烯在未来电子器件方面具有潜在的应用前景.
4.5 “自然图案化”的新型二维原子晶体材料及其功能化
一般来讲,二维原子晶体材料需要进行功能化或图案化才可能实现进一步的应用.例如,在半导体产业中,半导体材料需要先利用光刻技术图案化,再进行电子掺杂或空穴掺杂,进而形成P-N结和晶体管等.二维原子晶体材料的功能化可以通过多种方式实现,例如,将分子或原子沉积到二维原子晶体材料表面实现掺杂,或者引入另一种二维原子晶体材料构建叠层结构等[65].
基于这一思路,我们发展了一种构建纳米级精准规则图案的方法,首次构筑了两种基于过渡金属硫族化合物的纳米尺度的“自然图案化”材料:一种是具有交替三角形拼图图案的1H/1T型单层二硒化铂(1H/1T-PtSe2),另一种是具有周期排列三角形孔洞的单层硒化铜(CuSe).进一步将功能分子和磁性原子分别沉积到这两种材料表面的实验显示这两种二维原子晶体材料具有选择性功能化的特性[66].
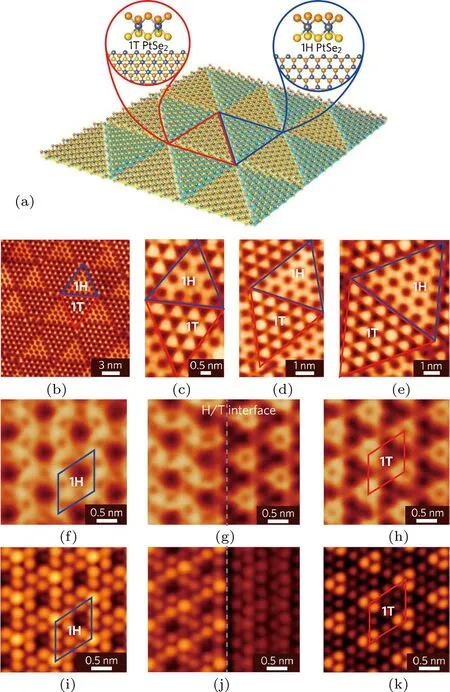
图21 具有交替三角形拼图图案的1H/1T型单层PtSe2[66](a)结构的示意图,分别标示出1H相和1T相的结构模型;(b)大面积STM图;(c)—(e)不同尺寸的1H相和1T相的STM图;(f)—(h)1H 相、1H/1T界面、1T相的高分辨STM图;(i)—(k)为(f)—(h)对应的STM模拟图Fig.21. 1H/1T tiling pattern in monolayer PtSe2[66]:(a)Schematic illustration of the triangular tiling pattern formed by alternating 1H and 1T PtSe2areas;(b)STM image of the 1H/1T patterned structure in monolayer PtSe2;(c)–(e)STM images of 1H and 1T PtSe2domains of di ff erent sizes;(f)–(h)high-resolution STM images of 1H PtSe2(f),1H/1T interface(g)and 1T PtSe2(h);the unit cells of the 1H and 1T domains are marked by blue and red diamonds,respectively;(i)–(k)STM simulations of a 1H PtSe2domain(i),the 1H/1T interface(j)and a 1T PtSe2 domain(k).
单层二硒化铂(PtSe2)具有两种构型,八面体型(1T)和三棱柱型(1H).2015年,我们利用直接硒化Pt(111)基底的方法,制备出了高质量半导体性质的1T型单层二硒化铂(1T-PtSe2)单晶薄膜[63].在此工作基础上,通过对1T型单层二硒化铂薄膜(1T-PtSe2)进行退火处理(基底400◦C),使其表面形成硒原子空位缺陷,空位缺陷重新排列得到了三角形图案化的1H/1T-PtSe2交替排列的单层薄膜,如图21所示.进一步对1H/1T-PtSe2薄膜补充硒原子并在较低温度下(基底270◦C)退火,可以获得纯的1T-PtSe2薄膜,从而实现了1H/1T-PtSe2薄膜和1T-PtSe2薄膜之间的可逆转变.将并五苯(Pentacene)分子沉积到1H/1T-PtSe2表面,发现并五苯分子选择性地吸附在三角形的1H-PtSe2区域,证实了该材料的选择性功能化特性.
在进一步的实验中,我们将硒原子沉积到Cu(111)单晶表面,首次成功构筑了新型类石墨烯二维原子晶体材料——硒化铜(CuSe),如图22所示.单层硒化铜(CuSe)具有蜂窝状结构,六元环中硒原子与铜原子交替排列成一种新型的双组分二维原子晶体材料,同时,为了释放CuSe与Cu(111)由于晶格失配产生的应力,单层硒化铜形成了一种自然图案化的二维原子晶体材料——六角周期排列的三角形纳米孔洞结构,孔洞的形状是边长为1 nm的等边三角形;结合密度泛函理论计算发现,只有周期性地缺失3个相邻的CuSe六元环才能够完全释放CuSe与Cu(111)由于晶格失配产生的应力,从而形成了周期性的三角形孔洞阵列.将铁原子(Fe)和酞菁铁分子(FePc)分别沉积到单层硒化铜表面,发现Fe原子仅在三角形孔洞内部形成Fe13Se7团簇,而FePc分子则会选择性吸附在非孔洞区域,证实了该自然图案化CuSe结构与1H/1T-PtSe2类似,同样具有选择性功能化的特性.
1H/1T-PtSe2和CuSe这两种具有纳米尺度自然图案的新型二维原子晶体材料虽然在超高真空环境中获得,它们在空气中也有很好的稳定性,预示了两种材料潜在的功能化应用前景.
这一工作发表在《Nature Materials》上[66].国际知名学者Joseph W.Lyding教授在同期的“News and Views(新闻和观点)”栏目上以“基于硫族化合物的二维材料——纳米尺度自然图案(Chalcogenide-based 2D materials:Intrinsic nanoscale patterning)”为题对此工作给予了高度评价,指出:“在制备PtSe2和CuSe单层结构时,发展了一种构建纳米级精准规则图案的方法(A method to realize regular patterns with nanometre precision during the synthesis of PtSe2and CuSe monolayers has been developed)”,“若将高鸿钧及其团队成员证实的自然图案化方法推广到一大类以硫族化合物为主的二维材料中,会为制备纳米尺寸器件及化学过程系统创造更多的机会(The generalization to a very broad class of chalcogenidebased 2D materials will create further opportunities for fabricating nanoscale devices and chemical processing systems using the intrinsic nanoscale patterning processes demonstrated by Gao and colleagues)”.
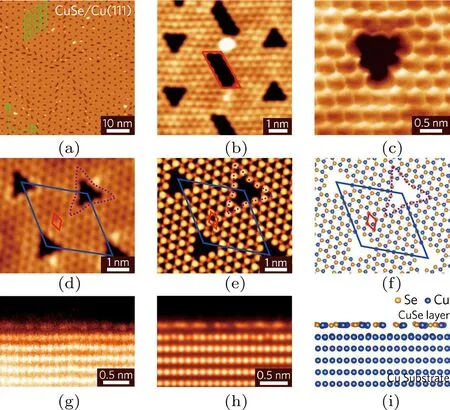
图22 具有周期排列三角形孔洞的单层CuSe[66] (a)大面积STM图,绿色格子标出周期为3 nm的规则孔洞;(b)畴界上的高分辨STM图显示,畴界由一些平行四边形的孔洞构成;(c)孔洞的高分辨STM 图,可见CuSe的六角蜂房形格子和在孔洞处的zigzag边界;(d)CuSe薄膜的原子分辨STM图,可见周期排列的三角形孔洞结构;(e)STM模拟图;(f)优化后的原子模型顶视图;(g)剖面STEM图;(h)STEM模拟图;(i)优化后的原子模型侧视图Fig.22. Patterned CuSemonolayerwith periodic nanopores[66]: (a)A large-scale STM image of patterned CuSe monolayer;the green lattice presents the 3 nm periodicity of the nanopores;(b)a high-resolution STM image of a domain-boundary region;the boundary consists of parallelogram-shaped nanopores as indicated by the red parallelogram;(c)a high-resolution STM image of a single nanopore;honeycomb lattice of CuSe and the zigzag edges of the nanopore are clearly resolved;(d)–(f)an atomicresolution STM image(d),simulated STM image(e),and theoretical model(f)of CuSe monolayer on Cu(111)with periodic nanopores;the images show the detailed structure of nanopores;in(f)Cu substrate is hidden for visualization purposes;(g)–(i)cross-section STEM image(g),simulated cross-section STEM image(h),and side view(i)of a CuSe monolayer on a Cu(111)substrate;the STEM image confi rms that the patterned CuSe is single layer.
本节介绍了石墨烯及几种新型二维原子晶体材料的可控构筑和物性调控,包括晶圆尺寸、高质量、单晶石墨烯的制备及其原位硅插层绝缘化,在多种基底上成功构筑了硅烯和锗烯,制备d电子金属元素构成的二维蜂窝状材料铪烯和V族元素二维蜂窝状材料锑烯.在此基础上,发展了一种构筑纳米级精确的“自然图案化”的新型二维原子晶体材料的方法,成功地制备出具有交替三角形拼图图案的1H/1T-PtSe2和具有周期排列三角形孔洞的单层CuSe,为构筑纳米尺寸器件提供了新的思路.这些材料体系多数为国际上首次创制,相关工作在国际上居引领地位.这些新型二维原子晶体材料的成功构筑不仅丰富了二维原子晶体材料库,同时为探索其物理特性和潜在的应用奠定了基础.
5 结 论
本文介绍了高鸿钧课题组在物理所20年来的部分代表性工作.研究的主要方向为低维纳米功能材料的分子束外延可控制备、生长机制、物性调控及其在未来信息技术中的原理性应用.
在材料的调控生长与机制研究方面,阐明了纳米尺度“海马”分形结构的形成及其生长机制;实现了通过对STM针尖进行修饰而获得表面纳米结构中更加精细的电子结构信息,并得到了Si(111)-7×7表面最高分辨的STM图像;证明了Ge原子在Si(111)表面的初期吸附时存在Ge替代Si的吸附机制,解决了之前20多年间Ge在Si(111)-7×7表面上初期吸附位置这一在理论和实验上一直悬而未决的问题;研究了多种分子在固体表面的吸附和自组装行为;提出了利用功能基团实现分子的非模板选择性自组装的方法.
在纳米量子结构电导和单个自旋态的可逆操纵及其在超高密度信息存储的原理性方面,利用具有双稳态结构的有机分子在外场作用下自身电导的改变,实现稳定、重复、可逆的超高密度信息存储;阐明了通过控制分子在表面的吸附构型和修饰分子中心的磁性原子两种方法来调控单个磁性分子自旋态的方法;通过STM分子“手术”的方法得到了自旋轨道耦合作用在单分子尺度的空间分布信息,提出了朗德g因子在单分子内部具有不均匀的空间分布.
在新型二维原子晶体的探索制备与物性研究方面,成功制备了多种二维原子晶体材料.其中包括在单晶金属上制备了晶圆尺寸、高质量、单晶石墨烯和构筑异质结结构,硅烯、锗烯、铪烯和锑烯的构筑,阐明了它们的生长机制,研究了它们的基本物性.更重要的是,发展了一种制备纳米级精确“自然图案化”的新型二维原子晶体材料的方法,成功地制备出具有交替三角形拼图图案的1H/1T-PtSe2和具有周期排列三角形孔洞的单层CuSe,为制备纳米尺寸器件及其功能化提供了新的思路.
这些工作阐明了低维纳米功能材料在可控构筑及物性调控中的基本物理问题,提出了材料功能化的新方法,奠定了这一类材料在未来电子器件应用及新量子现象探索研究中的基础.
在已有工作的基础上,我们认为低维纳米功能材料在未来发展主要包括以下四个方面:1)新型二维原子/分子晶体的设计、构筑及其机制;2)材料及其异质结结构与物性调控的研究;3)原理性功能器件的实现研究;4)实用化器件构筑与集成器件的研究.利用低维纳米功能材料实现超强的功能,引领该领域的科学前沿,以及催生未来技术,需要长期的、多学科和交叉领域科学家们不懈的努力.
感谢北京大学吴全德院士、薛增泉教授和中国科学院物理研究所庞世谨研究员早期的指导与支持;感谢曾经在实验室工作过的时东霞研究员、张秀芳高级工程师、张昊旭博士、刘虹雯博士、徐明春博士、肖文德博士等;感谢所有在实验室工作过的学生和博士后;感谢中国科学院化学研究所朱道本院士、刘云圻院士、张德清研究员、宋延林研究员,南开大学陈永胜教授,香港科技大学唐本忠院士等国内的合作者;感谢美国Vanderbilt大学S.T.Pantelides教授、Maryland大学M.Ouyang教授、Utah大学的Feng Liu教授、Purdue大学Y.Chen教授、橡树岭国家实验室S.J.Pennycook教授、英国Newcastle大学W.Hofer教授、德国Muenster大学H.Fuchs教授、德国马普所FHI H.J.Freund教授、Dresden工业大学X.L.Feng教授、美国加州大学Irvine分校W.Ho教授、瑞士EMPA研究所K.H.Ernst教授和Zurich大学T.Greber教授的合作;感谢所有合作者.
[1]Sato A,Tsukamoto Y 1993 Nature 363 431
[2]Crone B,Dodabalapur A,Lin Y Y,Filas R W,Bao Z,LaDuca A,Sarpeshkar R,Katz H E,Li W 2000 Nature 403 521
[3]Dimitrakopoulos C D,Malenfant P R L 2002 Adv.Mater.14 99
[4]Geim A K,Novoselov K S 2007 Nat.Mater.6 183
[5]Castro Neto A H,Guinea F,Peres N M R,Novoselov K S,Geim A K 2009 Rev.Mod.Phys.81 109
[6]Gao H J,Xue Z Q,Wu Q D,Pang S 1994 J.Mater.Res.9 2216
[7]Gao H J,Xue Z Q,Pang S J 1997 Thin Solid Films 292 40
[8]Cai J M,Bao L H,Guo W,Cai L,Huan Q,Lian J C,Guo H M,Wang K Z,Shi D X,Pang S J,Gao H J 2007 Chin.Phys.Lett.24 2918
[9]Gao H J,Zhang H X,Xue Z Q,Pang S J 1997 J.Mater.Res.12 1942
[10]Sandler I M,Canright G S,Gao H J,Pang S J,Xue Z Q,Zhang Z Y 1998 Phys.Rev.E 58 6015
[11]Sandler I M,Canright G S,Zhang Z Y,Gao H J,Xue Z Q,Pang S J 1998 Phys.Lett.A 245 233
[12]Hamers R J,Tromp R M,Demuth J E 1986 Phys.Rev.Lett.56 1972
[13]Giessibl F J 1995 Science 267 68
[14]Wang Y L,Gao H J,Guo H M,Liu H W,Batyrev I G,McMahon W E,Zhang S B 2004 Phys.Rev.B 70 073312
[15]Cheng Z H,Du S X,Guo W,Gao L,Deng Z T,Jiang N,Guo H M,Tang H,Gao H J 2011 Nano Res.4 523
[16]Cheng Z H,Du S X,Jiang N,Zhang Y Y,Guo W,Hofer W A,Gao H J 2011 Surf.Sci.605 415
[17]Deng Z T,Lin H,Ji W,Gao L,Lin X,Cheng Z H,He X B,Lu J L,Shi D X,Hofer W A,Gao H J 2006 Phys.Rev.Lett.96 156102
[18]Wang Y L,Gao H J,Guo H M,Wang S,Pantelides S T 2005 Phys.Rev.Lett.94 106101
[19]Shi D X,Ji W,Lin X,He X B,Lian J C,Gao L,Cai J M,Lin H,Du S X,Lin F,Seidel C,Chi L F,Hofer W A,Fuchs H,Gao H J 2006 Phys.Rev.Lett.96 226101
[20]Jiang N,Zhang Y Y,Liu Q,Cheng Z H,Deng Z T,Du S X,Gao H J,Beck M J,Pantelides S T 2010 Nano Lett.10 1184
[21]Jiang N,Wang Y,Liu Q,Zhang Y,Deng Z,Ernst K H,Gao H J 2010 Phys.Chem.Chem.Phys.12 1318
[22]Yang B,Wang Y L,Li G,Cun H Y,Ma Y,Du S X,Xu M C,Song Y L,Gao H J 2009 J.Phys.Chem.C 113 17590
[23]Yang B,Wang Y,Cun H,Du S,Xu M,Wang Y,Ernst K H,Gao H J 2010 J.Am.Chem.Soc.132 10440
[24]Cun H,Wang Y,Yang B,Zhang L,Du S,Wang Y,Ernst K H,Gao H J 2010 Langmuir 26 3402
[25]Cun H,Wang Y,Du S,Zhang L,Zhang L,Yang B,He X,Wang Y,Zhu X,Yuan Q,Zhao Y P,Ouyang M,Hofer W A,Pennycook S J,Gao H J 2012 Nano Lett.12 1229
[26]Cheng Z H,Gao L,Deng Z T,Jiang N,Liu Q,Shi D X,Du S X,Guo H M,Gao H J 2007 J.Phys.Chem.C 111 9240
[27]Cheng Z H,Gao L,Deng Z T,Liu Q,Jiang N,Lin X,He X B,Du S X,Gao H J 2007 J.Phys.Chem.C 111 2656
[28]Burke S,Ji W,Mativetsky J,Topple J,Fostner S,Gao H J,Guo H,Grütter P 2008 Phys.Rev.Lett.100 186104
[29]Hu H,Gao H J,Liu F 2008 Phys.Rev.Lett.101 216102
[30]Du S X,Gao H J,Seidel C,Tsetseris L,Ji W,Kopf H,Chi L F,Fuchs H,Pennycook S J,Pantelides S T 2006 Phys.Rev.Lett.97 156105
[31]Eremtchenko M,Schaefer J A,Tautz F S 2003 Nature 425 602
[32]Hauschild A,Karki K,Cowie B C C,Rohl fi ng M,Tautz F S,Sokolowski M 2005 Phys.Rev.Lett.94 036106
[33]Wen Y Q,Song Y L,Jiang G Y,Zhao D B,Ding K L,Yuan W F,Lin X,Gao H J,Jiang L,Zhu D B 2004 Adv.Mater.16 2018
[34]Wen Y Q,Wang J X,Hu J P,Jiang L,Gao H J,Song Y L,Zhu D B 2006 Adv.Mater.18 1983
[35]Jiang G Y,Michinobu T,Yuan W F,Teng M,Wen Y Q,Du S X,Gao H J,Jiang L,Song Y L,Diederich F,Zhu D B 2005 Adv.Mater.17 2170
[36]Ma Y,Cao X,Li G,Wen Y,Yang Y,Wang J,Du S,Yang L,Gao H J,Song Y 2010 Adv.Funct.Mater.20 803
[37]Wu H M,Song Y L,Du S X,Liu H W,Gao H J,Jiang L,Zhu D B 2003 Adv.Mater.15 1925
[38]Hu Y B,Zhu Y,Gao H J,Guo H 2005 Phys.Rev.Lett.95 156803
[39]Gao H J,Gao L 2010 Prog.Surf.Sci.85 28
[40]Feng M,Guo X F,Lin X,He X B,Ji W,Du S X,Zhang D Q,Zhu D B,Gao H J 2005 J.Am.Chem.Soc.127 15338
[41]Feng M,Gao L,Deng Z T,Ji W,Guo X F,Du S X,Shi D X,Zhang D Q,Zhu D B,Gao H J 2007 J.Am.Chem.Soc.129 2204
[42]Feng M,Gao L,Du S X,Deng Z T,Cheng Z H,Ji W,Zhang D Q,Guo X F,Lin X,Chi L F,Zhu D B,Fuchs H,Gao H J 2007 Adv.Funct.Mater.17 770
[43]Gao L,Ji W,Hu Y B,Cheng Z H,Deng Z T,Liu Q,Jiang N,Lin X,Guo W,Du S X,Hofer W A,Xie X C,Gao H J 2007 Phys.Rev.Lett.99 106402
[44]Ren J,Guo H,Pan J,Zhang Y Y,Wu X,Luo H G,Du S,Pantelides S T,Gao H J 2014 Nano Lett.14 4011
[45]Ren J D,Guo H M,Pan J,Zhang Y F,Yang Y,Wu X,Du S,Ouyang M,Gao H J 2017 Phys.Rev.Lett.119 176806
[46]Gao L,Liu Q,Zhang Y Y,Jiang N,Zhang H G,Cheng Z H,Qiu W F,Du S X,Liu Y Q,Hofer W A,Gao H J 2008 Phys.Rev.Lett.101 197209
[47]Liu Q,Zhang Y Y,Jiang N,Zhang H G,Gao L,Du S X,Gao H J 2010 Phys.Rev.Lett.104 166101
[48]Liu Q,Du S,Zhang Y,Jiang N,Shi D,Gao H J 2012 Small 8 796
[49]Yang K,Liu L,Zhang L,Xiao W,Fei X,Chen H,Du S,Ernst K H,Gao H J 2014 ACS Nano 8 2246
[50]Liu L W,Yang K,Jiang Y H,Song B Q,Xiao W D,Li L F,Zhou H T,Wang Y L,Du S X,Ouyang M,Hofer W A,Castro Neto A H,Gao H J 2013 Sci.Rep.3 1210
[51]Liu L W,Yang K,Jiang Y H,Song B Q,Xiao W D,Song S R,Du S X,Ouyang M,Hofer W A,Castro Neto A H,Gao H J 2015 Phys.Rev.Lett.114 126601
[52]Novoselov K S,Geim A K,Morozov S V,Jiang D,Zhang Y,Dubonos S V,Grigorieva I V,Firsov A A 2004 Science 306 666
[53]Pan Y,Shi D X,Gao H J 2007 Chin.Phys.16 3151
[54]Pan Y,Zhang H G,Shi D X,Sun J T,Du S X,Liu F,Gao H J 2009 Adv.Mater.21 2777
[55]Gao M,Pan Y,Huang L,Hu H,Zhang L Z,Guo H M,Du S X,Gao H J 2011 Appl.Phys.Lett.98 033101
[56]Meng L,Wu R T,Zhang L Z,Li L F,Du S X,Wang Y L,Gao H J 2012 J.Phys.:Condens.Matter 24 314214[57]Pan Y,Zhang L,Huang L,Li L,Meng L,Gao M,Huan Q,Lin X,Wang Y,Du S,Freund H J,Gao H J 2014 Small 10 2215
[58]Meng L,Wang Y L,Zhang L Z,Du S X,Wu R T,Li L,Zhang Y,Li G,Zhou H T,Hofer W A,Gao H J 2013 Nano Lett.13 685
[59]Huang L,Zhang Y F,Zhang Y Y,Xu W Y,Que Y D,Li E,Pan J B,Wang Y L,Liu Y Q,Du S X,Pantelides S T,Gao H J 2017 Nano Lett.17 1161
[60]Li L F,Lu S Z,Pan J B,Qin Z H,Wang Y Q,Wang Y L,Cao G Y,Du S X,Gao H J 2014 Adv.Mater.26 4820
[61]Qin Z,Pan J,Lu S,Shao Y,Wang Y,Du S,Gao H J,Cao G 2017 Adv.Mater.29 1606046
[62]Li L F,Wang Y L,Xie S,Li X B,Wang Y Q,Wu R T,Sun H B,Zhang S B,Gao H J 2013 Nano Lett.13 4671
[63]Wang Y L,Li L F,Yao W,Song S R,Sun J T,Pan J B,Ren X,Li C,Okunishi E,Wang Y Q,Wang E,Shao Y,Zhang Y Y,Yang H T,Schwier E F,Iwasawa H,Shimada K,Taniguchi M,Cheng Z H,Zhou S Y,Du S X,Pennycook S J,Pantelides S T,Gao H J 2015 Nano Lett.15 4013
[64]Wu X,Shao Y,Liu H,Feng Z,Wang Y L,Sun J T,Liu C,Wang J O,Liu Z L,Zhu S Y,Wang Y Q,Du S X,Shi Y G,Ibrahim K,Gao H J 2017 Adv.Mater.29 1605407
[65]Wang Y Q,Wu X,Wang Y L,Shao Y,Lei T,Wang J O,Zhu S Y,Guo H,Zhao L X,Chen G F,Nie S,Weng H M,Ibrahim K,Dai X,Fang Z,Gao H J 2016 Adv.Mater.28 5013
[66]Lin X,Lu J C,Shao Y,Zhang Y Y,Wu X,Pan J B,Gao L,Zhu S Y,Qian K,Zhang Y F,Bao D L,Li L F,Wang Y Q,Liu Z L,Sun J T,Lei T,Liu C,Wang J O,Ibrahim K,Leonard D N,Zhou W,Guo H M,Wang Y L,Du S X,Pantelides S T,Gao H J 2017 Nat.Mater.16 717
[67]Rutter G M,Crain J N,Guisinger N P,Li T,First P N,Stroscio J A 2007 Science 317 219
[68]Li X,Cai W,An J,Kim S,Nah J,Yang D,Piner R,Velamakanni A,Jung I,Tutuc E,Banerjee S K,Colombo L,Ruo ffR S 2009 Science 324 1312
[69]Sutter P W,Flege J I,Sutter E A 2008 Nat.Mater.7 406
[70]Mao J H,Huang L,Pan Y,Gao M,He J F,Zhou H T,Guo H M,Tian Y,Zou Q,Zhang L Z,Zhang H G,Wang Y L,Du S X,Zhou X J,Neto A H C,Gao H J 2012 Appl.Phys.Lett.100 093101
[71]Meng L,Wu R T,Zhou H T,Li G,Zhang Y,Li L L,Wang Y L,Gao H J 2012 Appl.Phys.Lett.100 083101
[72]Que Y D,Zhang Y,Wang Y L,Huang L,Xu W Y,Tao J,Wu L J,Zhu Y M,Kim K,Weinl M,Schreck M,Shen C M,Du S X,Liu Y Q,Gao H J 2015 Adv.Mater.Inter.2 1400543
[73]Li G,Zhou H,Pan L,Zhang Y,Huang L,Xu W,Du S,Ouyang M,Ferrari A C,Gao H J 2015 J.Am.Chem.Soc.137 7099
[74]Gao M,Pan Y,Zhang C D,Hu H,Yang R,Lu H L,Cai J M,Du S X,Liu F,Gao H J 2010 Appl.Phys.Lett.96 053109
[75]Huang L,Pan Y,Pan L D,Gao M,Xu W Y,Que Y D,Zhou H T,Wang Y L,Du S X,Gao H J 2011 Appl.Phys.Lett.99 163107
[76]Fei X M,Zhang L Z,Xiao W D,Chen H,Que Y D,Liu L W,Yang K,Du S X,Gao H J 2015 J.Phys.Chem.C 119 9839
[77]Li L,Wang Y,Meng L,Wu R T,Gao H J 2013 Appl.Phys.Lett.102 093106
[78]Guo H,Lu H L,Huang L,Wang X Y,Lin X,Wang Y L,Du S X,Gao H J 2017 Acta Phys.Sin.66 216803(in Chinese)[郭辉,路红亮,黄立,王雪艳,林晓,王业亮,杜世萱,高鸿钧2017物理学报66 216803]
[79]Xu W Y,Huang L,Que Y D,Lin X,Wang Y L,Du S X,Gao H J 2014 Chin.Phys.B 23 088108
[80]Li G,Zhou H T,Pan L D,Zhang Y,Mao J H,Zou Q,Guo H M,Wang Y L,Du S X,Gao H J 2012 Appl.Phys.Lett.100 013304
[81]Yang K,Xiao W D,Jiang Y H,Zhang H G,Liu L W,Mao J H,Zhou H T,Du S X,Gao H J 2012 J.Phys.Chem.C 116 14052
[82]Mao J H,Zhang H G,Jiang Y H,Pan Y,Gao M,Xiao W D,Gao H J 2009 J.Am.Chem.Soc.131 14136
[83]Zhou H T,Mao J H,Li G,Wang Y L,Feng X L,Du S X,Müllen K,Gao H J 2011 Appl.Phys.Lett.99 153101[84]Zhang H G,Sun J T,Low T,Zhang L Z,Pan Y,Liu Q,Mao J H,Zhou H T,Guo H M,Du S X,Guinea F,Gao H J 2011 Phys.Rev.B 84 245436
[85]Zhang H G,Xiao W D,Mao J H,Zhou H T,Li G,Zhang Y,Liu L W,Du S X,Gao H J 2012 J.Phys.Chem.C 116 11091
[86]Fei X M,Neilson J,Li Y B,Lopez V,Garrett S J,Gan L B,Gao H J,Gao L 2017 Nano Lett.17 2887
[87]Pan L D,Que Y D,Chen H,Wang D F,Li J,Shen C M,Xiao W D,Du S X,Gao H J,Pantelides S T 2015 Nano Lett.15 6464
[88]Que Y D,Xiao W D,Fei X M,Chen H,Huang L,Du S X,Gao H J 2014 Appl.Phys.Lett.104 093110
猜你喜欢
昆钢科技(2022年1期)2022-04-19
中国银幕(2022年4期)2022-04-07
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
纺织科学研究(2021年7期)2021-08-14
中学数学研究(广东)(2018年23期)2018-03-05
山东工业技术(2016年15期)2016-12-01
通信电源技术(2016年6期)2016-04-20
中国卫生标准管理(2015年7期)2015-01-27
