
二维液相色谱法在线富集测定银杏叶提取物中银杏酸含量
2018-07-06 01:53杜小弟李俊升郭丽萍雷家珩
分析测试学报 2018年6期
杜小弟,李俊升,郭丽萍,雷家珩
(武汉理工大学 化学化工与生命科学学院 化学系,湖北 武汉 430070)

图1 银杏酸的结构式Fig.1 Structural formula of Ginkgolic acids
银杏是中国特有的药用植物,在延缓衰老、改善血液循环、治疗心脑血管疾病等方面有一定疗效[1-3]。但临床发现银杏可导致接触过敏性皮炎,并存在潜在的细胞毒性、神经毒性等副作用[4-5]。研究认为,银杏酸(Ginkgolic acids,GAs)是引起副作用的重要原因[6]。较为常见的银杏酸包括GA13∶0、GA15∶1、 GA17∶2、GA15∶0和GA17∶1,其结构如图1所示。
对于药用标准的银杏叶提取物(Extracts of Ginkgo biloba,EGB),国外药典不仅规定了其有效成分的含量,还限定了总银杏酸的含量不得超过5 mg/kg[7-8],我国药典也有类似规定[9]。因此,需要建立一种检测EGB及其制剂中银杏酸含量的简便、可靠、灵敏的方法。EGB中的银杏酸可以通过液相色谱(HPLC)、气相色谱(GC)、薄层色谱(TLC)等方法进行测定[10-11]。但薄层色谱的准确度和灵敏度均不高,气相色谱法需衍生化处理[12],仅液相色谱法应用最为广泛。Fuzzati等[13]开发的提取液直接进样、液相色谱紫外检测器(HPLC/UV)测定银杏酸的方法由于简便性突出,且经过了完善的方法学确认,被采纳为欧盟药典中检测EGB的标准方法[7]。然而该方法存在以下不足:①不经分离直接进样,虽然方法简便,但基体干扰较为显著;②为提高灵敏度而使用较大的进样量(50 μL)和较低的检测波长(210 nm),使得基体干扰和基线噪音问题更加突出,因此该方法在应用中仍需改进[14-15]。而且该方法的检出限仅能够满足EGB中银杏酸的限量检测,对于含量更低的EGB制剂(如银杏叶片、银杏叶滴丸等)则难以测定。为提高测定的灵敏度、减小复杂基体的干扰,研究者尝试使用液液萃取(LLE)[16]、固相萃取(SPE)[17]等方法以降低基体干扰,但操作较为繁琐、耗时长。也有采用超高效液相色谱-串联质谱联用(UHPLC-MS/MS)测定微量银杏酸的方法[18],但仪器昂贵不便推广。
近年来,在线二维色谱在中药成分研究方面得到了广泛应用[19],以此为基础的在线分离富集方法已有一定的研究[20-21]。有学者通过全二维色谱对银杏成分进行定性研究[22],但未进行定量分析。有文献通过双柱切换的方法测定银杏叶中的银杏酸含量[23],在消除基体干扰方面有一定效果,但未能实现富集,难以适用于微量银杏酸的测定。为减少基体干扰,同时实现微量银杏酸的富集,本文搭建了在线二维色谱系统,将大体积的试样通过第一维色谱分离实现富集和除去基体干扰,然后通过阀切换直接将目标物引入第二维色谱分离进行定量检测,从而实现EGB及其制剂中微量银杏酸的准确测定。

图2 二维色谱系统的构成和阀切换示意图Fig.2 Schematic of the two-dimensional liquid chromatography system and the valve switching
1 实验部分
1.1 仪器、试剂与材料
在线二维色谱系统由2台LC-20AD高压恒流泵(日本岛津公司)、1台SPD-20A紫外检测器(日本岛津公司)、1个7725i手动进样阀(配300 μL定量环,美国Rheodyne公司)、1个7000型高压切换阀(美国Rheodyne公司)和2根色谱柱组成,示意图见图2。其中柱1为ZORBAX Eclipse Plus-C18(2.1 mm×12.5 mm,5 μm),柱2为ZORBAX Eclipse Plus-C18(2.1 mm×150 mm,5 μm),均购于美国安捷伦公司。进样阀、切换阀和色谱柱均安装在CTO-10ASvp柱温箱(日本岛津公司)内。
EGB试样为湖北盛天恒创公司生产,EGB制剂试样为市售银杏叶片药物(金纳多)。混合银杏酸标准品购于武汉天植生物技术公司,总含量不低于99%,其中GA13∶0占10.1%,GA15∶1占50.5%,GA17∶1占34.7%,GA17∶2占1.7%,GA15∶0占3.0%。用甲醇溶解配制成总银杏酸质量浓度为1.000 g/L的标准储备液,4 ℃保存。使用时以甲醇-水-三氟乙酸混合液(体积比80∶20∶0.01)稀释成所需浓度的工作标液。
甲醇、乙腈为色谱纯(国药集团化学试剂有限公司),三氟乙酸(纯度99%,阿拉丁试剂上海有限公司),实验用水为二次蒸馏水。
1.2 色谱条件
泵1和柱1为第一维分离,流动相为甲醇-水-三氟乙酸混合液(体积比80∶20∶0.01),流速0.25 mL/min。泵2和柱2为第二维分离,流动相为甲醇-水-三氟乙酸混合液(体积比90∶10∶0.01),流速0.25 mL/min。柱温均为30 ℃。紫外检测器波长为310 nm。进样量为300 μL(300 μL定量环全充满)。切换阀处于图2A所示状态时,进样4.5 min,后将切换阀切换至图2B所示状态。测定结束后切换回图2A状态,平衡2 min后进行下次测定。
1.3 实验方法
样品测定:准确称取EGB样品(或粉碎后的制剂样品)0.500 g置于10 mL比色管中,加入甲醇8.0 mL,常温下超声提取10 min,加水定容至10.0 mL,用微量注射器加入三氟乙酸1.0 μL。混匀后用0.45 μm尼龙滤膜过滤,收集滤液,按“1.2”所述色谱条件进行测定。
样品加标回收实验:准确称取EGB样品(或粉碎后的制剂样品)0.500 g置于10 mL比色管中,准确加入总银杏酸质量浓度为0.100 g/L的工作标液25.0 μL(相当于样品加标5 mg/kg)或50.0 μL(相当于样品加标10 mg/kg),后续操作与样品测定相同。
1.4 对照方法
参照欧洲药典EP8.0所述方法[8]:准确称取EGB样品0.500 g,用10.0 mL甲醇超声提取,提取液过滤后直接进行色谱测定。色谱柱为Zorbax XDB-C8(4.6 mm×150 mm,5 μm,美国安捷伦公司),流动相:A为0.01% 三氟乙酸乙腈溶液,B为0.01%三氟乙酸水溶液;梯度洗脱程序:初始为75%A,30 min线性改变至90%A;流速1.00 mL/min,检测波长210 nm,进样量50.0 μL。
2 结果与讨论
2.1 实验条件考察
2.1.1二维色谱系统的构建在图2所示的二维色谱系统中,柱1进行第一维分离,使目标物与大部分基体干扰物分开。同时,通过大体积进样,使目标物被吸附在柱1上,从而实现富集。通过阀切换将柱1接入第二维,通过第二维的流动相将柱1上富集的目标物洗脱,并在柱2上实现进一步的分离和定量检测。第二维中的柱2为分析柱,参考文献[14-19]中银杏酸的测定方法,选择十八烷基(C18)固定相,流动相为甲醇-水-三氟乙酸混合液(体积比90∶10∶0.01)。选择2.1 mm内径的窄内径色谱柱,在相同的进样体积下,获得的灵敏度为4.6 mm标准内径柱的4~5倍。
第一维中柱1除了达到预分离基体的目的外,还需对大体积样品中的目标物进行富集,因此需对目标物有较强的保留能力,比较了十八烷基(C18)、辛烷基(C8)、氰基(CN)3种键合固定相的效果。C8和CN固定相对银杏酸的保留能力较弱,富集效果差,需使用50 mm以上的长柱才能完全吸附大体积样品中的目标物,但长柱导致目标物洗脱速度慢,峰展宽严重;而C18固定相对银杏酸的保留较强,使用12.5 mm的短柱即可实现数百微升试样中目标物的富集,且短柱可以被快速洗脱,峰形尖锐。
2.1.2第一维色谱条件的选择第一维分离需要在柱1上将基体与目标物初步分离。为考察分离效果,分别测定了试样溶液和标液在柱1上流出的色谱图。当第一维以甲醇-水-三氟乙酸混合液(体积比80∶20∶0.01)为流动相时,所得色谱图见图3。此条件下,基体主要成分的流出时间约在4 min内,目标物银杏酸的流出时间约在5 min后,基本可实现分离。当甲醇-水体积比降至70∶30时,虽然目标物的流出时间更长,但基体的洗脱相应变慢,且峰形拖尾更明显,分离效果未改善。当甲醇-水体积比增至90∶10时,目标物流出太快,无法实现分离。


图3 银杏酸标样(A)与EGB试样基体(B)在第一维上的色谱图Fig.3 Chromatograms of GAs standard(A) and EGB sample matrix(B) on the first dimension
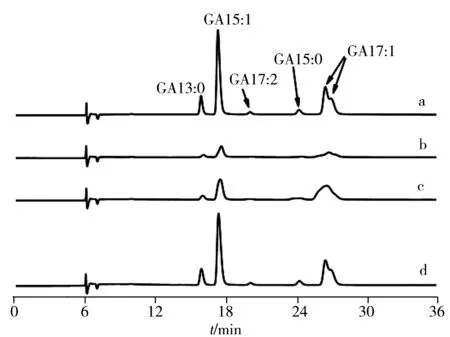
图4 样品溶剂对色谱结果的影响Fig.4 Effect of solvent on chromatograms 3 μL injection of GAs standard sample(25.0 mg/L) with the solvent of methanol(a),and 300 μL injection of GAs standard sample(0.250 mg/L) in the solvent of absolute methanol(b),90% methanol(c),80% methanol(d)
根据图3确定六通阀的切换时间为4.50 min。若切换时间提前,则会引入较多的基体组分,对第二维分离不利。若切换时间大于5.00 min,将导致目标物损失,使回收率显著降低。
2.1.3试样溶剂及进样体积对富集效果的影响2.1 mm内径的色谱柱进样量通常不超过5 μL。为实现富集,第一维须大体积进样。由于进样体积很大时溶剂的洗脱作用会较为显著,从而影响色谱结果。以甲醇为溶剂配制的GAs标样(25.0 mg/L)在进样体积为3 μL时,获得的色谱图见图4a,其中除GA17∶1由于异构体的存在表现为双峰外,其余4种目标物均具有尖锐的峰形。当使用GAs标样(0.250 mg/L,甲醇溶剂)进样300 μL时,由于溶剂甲醇的强洗脱作用,导致目标物在阀切换之前被洗脱,各目标物明显损失,且峰宽显著增大(见图4b)。当溶剂中甲醇-水体积比为90∶10时(GAs浓度为0.250 mg/L),溶剂的洗脱能力仍比流动相强,富集效果不佳,峰宽较大(见图4c)。当溶剂中甲醇-水体积比为80∶20(GAs质量浓度为0.250 mg/L),即溶剂与流动相组成一致时,溶剂的影响基本可以忽略,峰面积与图4d接近,说明富集效果较好,灵敏度提高近100倍,而且峰形尖锐,与小体积进样时的分离效果一致。
虽然大体积进样可实现富集,但随着进样体积的增大,各目标物的峰形逐渐展宽。考察了半峰宽随进样体积的变化,结果表明当进样体积在300 μL以下时峰宽变化不大,当进样体积超过300 μL时峰宽增加较为明显。因此选择最佳进样体积为300 μL。
2.2 方法学考察
通过配制不同含量的模拟样考察了方法的线性范围,以各目标物的峰面积对其含量进行线性回归,结果见表1。5种银杏酸在0.200~100.0 mg/kg范围内均具有良好的线性关系,相关系数(r2)不低于0.998。配制一系列低浓度的银杏酸标样测定信噪比(S/N),分别以S/N=3和S/N=10计算检出限和定量下限(见表1)。5种银杏酸的检出限为0.02~0.06 mg/kg,定量下限为0.05~0.19 mg/kg。本方法不仅适用于EGB样品中银杏酸的测定,还适用于EGB制剂中更微量银杏酸含量的测定。

表1 5种银杏酸的线性关系、检出限及定量下限Table 1 Linear relationship,limits of detection(LODs) and limits of quantitation(LOQs) of 5 ginkgolic acids
*Y:peak area;X:mass concentration(mg/kg)
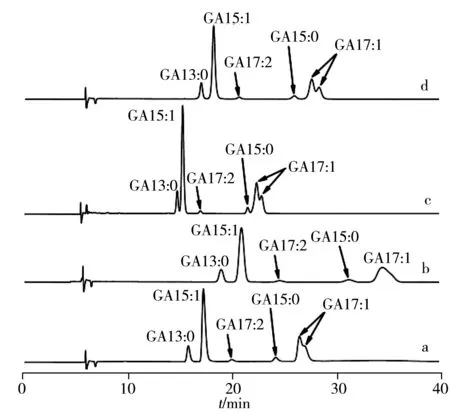
图5 本方法对不同色谱柱的适应性Fig.5 Adaptability of this method to different columnsa.ZORBAX Eclipse Plus-C18,b.XDB-C18,c.Hypersil GOLD-C18,d.SinoChrome ODS-BP
进一步对方法的适应性和耐用性进行了考察。除“1.1”所述的色谱柱外,另选用了不同厂家不同型号的3种色谱柱,包括安捷伦公司XDB-C18、Thermo公司Hypersil GOLD-C18、大连依利特公司SinoChrome ODS-BP(柱尺寸规格与“1.1”相同)。4种色谱柱的分离效果对比见图5。结果表明,不同型号的色谱柱虽对待测物的保留时间有所差异,但均能有效富集和分离,且分离效果和柱效均较理想,对GA13∶0的柱效不低于6 000理论塔板数,除GA17∶1的顺反异构体外,其余各银杏酸的分离度不低于1.5。实验进一步考察了色谱条件发生微小变化时的分离效果,结果表明,检测波长(±5 nm)、柱温(±10 ℃)、流速(±0.05 mL/min)的微小变化对测定效果影响不明显。以上实验表明本方法具有较好的适应性和耐用性。
2.3 实际样品检测
取市售EGB试样和EGB制剂试样按本方法进行处理和测定,以银杏酸纯品作为对照样进行定量,结果见表2。对照样的色谱图见图6a,EGB试样的色谱图见图6b。对于EGB试样,本方法具有很好的重现性,各目标物测定结果的相对标准偏差(RSD)均不超过5.0%,总银杏酸结果的RSD小于3.0%。对于EGB制剂,由于其中银杏酸含量较低,GA15∶0未检出,GA17∶2和GA17∶1能检出但低于定量下限,对于含量较高的GA13∶0和GA15∶1可有效测定且RSD不超过5.0%,总银杏酸结果的RSD为4.8%。

表2 EGB和EGB制剂试样中银杏酸含量的测定结果Table 2 Determination results of ginkgolic acids in EGB sample and EGB troche sample
*mean±SD,n=5;**mean±SD,n=5,measured by the control method described in Section“1.4”;***below the limit of quantitation

图6 本方法测定EGB试样和加标试样的色谱图Fig.6 Chromatograms of EGB sample and spiked EGB sample obtained by using proposed methoda.GAs standard sample(0.25 mg/L),b.EGB sample,c.spiked EGB sample at content level of 5 mg/kg for total GAs
考虑到药典对EGB中总银杏酸的限量为5 mg/kg[7-8]或10 mg/kg[9],而实际样品中的含量值一般与限量值接近,因此对EGB试样进行了总银杏酸为5、10 mg/kg两个浓度水平的加标实验,结果见表3。不同加标水平下,各目标物的回收率为94.0%~101.3%,总银杏酸的回收率接近100%,表明方法具有较高的准确度。总银杏酸5 mg/kg加标水平的色谱图见图6c。

图7 对照方法测定EGB试样的色谱图Fig.7 Chromatograms of EGB sample obtained by using the control methoda.GAs standard(0.25 mg/L),b.EGB sample
以欧盟药典方法[8]作为对照方法进行了对照实验,结果见表2。对于单一银杏酸,本方法测定结果与对照方法具有较好的一致性,且精密度优于对照方法。在灵敏度方面,对照方法的检出限约为0.3~0.5 mg/kg,而本方法的检出限约为对照方法的1/10,对于低含量的目标物GA17∶2和GA15∶0,本方法也可检出,而对照方法难以检出。因此,对于总银杏酸的计算结果,本方法略高于对照方法,若不计入GA17∶2和GA15∶0的含量,则本方法与对照方法结果一致。用对照方法测定EGB试样和对照样的色谱图见图7,本方法通过第一维分离有效消除了基体干扰,使色谱图基线更平直,干扰峰显著减少。而且本方法通过在线富集使灵敏度显著提高,采用更高的检测波长(310 nm)使噪音和基线漂移比对照方法(检测波长210 nm)显著降低,因此在相同浓度水平下,本方法的信噪比显著优于药典方法。对于EGB制剂中微量银杏酸的检测,本方法更有效。
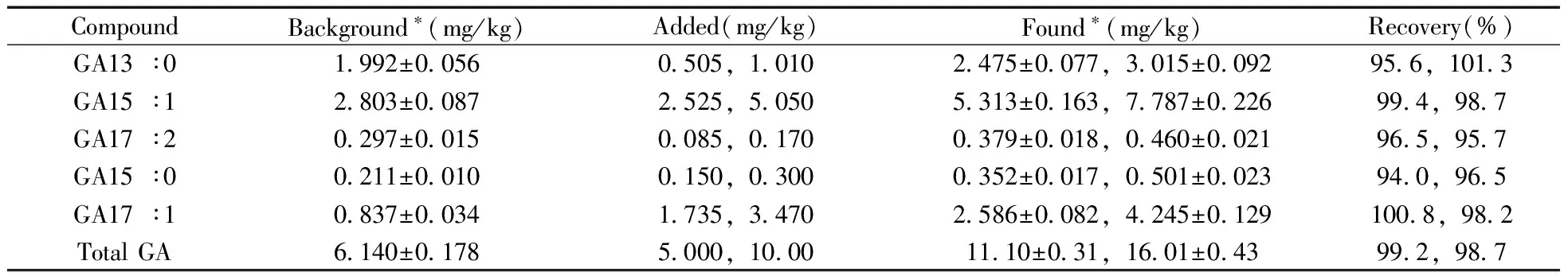
表3 EGB试样中银杏酸的加标回收率Table 3 Spiked recoveries of ginkgolic acids in EGB sample
*mean±SD,n=5
3 结 论
本文在自行搭建的二维液相色谱基础上,建立了在线富集测定EGB及其制剂中微量银杏酸的方法。该方法在有效消除基体干扰的同时实现了目标物的富集,检出限为现行药典方法的1/10。与LLE、SPE等离线富集方法相比,本方法的分离富集过程可通过阀切换自动完成,操作更简便,速度更快,由于操作带来的损失和不确定度更小。
参考文献:
[1] Elsabagh S,Hartley D E,File S E.J.Psychopharmacol.,2005,19(2):173-181.
[2] Wang S J,Chen H H.Eur.J.Pharmacol.,2005,514(2/3):141-149.
[3] Kalkunte S S,Singh A P,Chaves F C,Gianfagna T J,Pundir V S.PhytotherRes.,2010,21(11):1061-1065.
[4] Koch E,Noeldner M,Leuschner J.Phytomedicine,2013,21(1):90-97.
[5] Posadzki P,Watson L,Ernst E.Br.J.Clin.Pharmacol.,2013,75(3):603-618.
[6] Ahlemeyer B,Selke D,Schaper C,Klumpp S,Krieglstein J.Eur.J.Pharmacol.,2001,430(1):1-7.
[7] European Pharmacopoeia 8.0.Monograph:Ginkgo Dry Extract.Refined and Quantified.2008.European Directorate for the Quality of Medicines & Healthcare,Strasbourg,France.
[8] United States Pharmacopeia.Thirty-seventh Revision:Powdered Ginkgo Extract.2015.The United States Pharmacopeial Convention,Rockville,Md.
[9] Chinese Pharmacopoeia Commission.Pharmacopoeia of the People’s Republic of China,Part 1.Beijing:China Medical Science Press(国家药典委员会.中华人民共和国药典,一部.北京:中国医药科技出版社),2015:416-417.
[10] Teris A B.J.Chromatogr.A,2002,967(1):21-55.
[11] Teris A B,Paola M.J.Chromatogr.A,2009,1216(11):2002-2032.
[12] Sun Y,Tang C,Wu X,Pan Z,Wang L.Chromatographia,2012,75(7/8):387-395.
[13] Fuzzati N,Pace R,Villa F.Fitoterapia,2003,74(3):247-256.
[14] Yao J B,Jin H H,Wang R W,He H H,Zheng C.Chin.J.Pharm.Anal.(姚建标,金辉辉,王如伟,何厚洪,郑成.药物分析杂志),2015,35(11):2041-2044.
[15] Wang Q W,Xie Y Y,Wang Y M,Liang Q L,Luo G A.Chin.Pharm.J.(王荞薇,谢媛媛,王义明,梁琼麟,罗国安.中国药学杂志),2015,50(2):167-173.
[16] Stefan U,Iuliana D S,Victor D,Andrei M.J.Liq.Chromatogr.RT,2010,33(1):133-149.
[17] Ji W,Ma X,Xie H,Chen L,Wang X,Zhao H,Huang L.J.Chromatogr.A,2014,1368(1):44-51.
[18] Sun J,Li L M,Hu Q,Mao X H,Ji S.Chin.J.Chromatogr.(孙健,李丽敏,胡青,毛秀红,季申.色谱),2016,34(2):184-188.
[19] Gao W,Song H P,Yang H,Li P.Chin.J.Chromatogr.(高雯,宋慧鹏,杨华,李萍.色谱),2017,35(1):121-128.
[20] Liu Y L,Wang C S,Wang X L,Yang S N,Liu X G,Feng L.Chin.J.Anal.Chem.(刘永利,王常顺,王晓蕾,杨抒楠,刘兴国,冯丽.分析化学),2016,44(5):828-832.
[21] Zhang Y,Ma X F,Lü P,Cong B.Chin.J.Anal.Chem.(张岩,马晓斐,吕品,丛斌.分析化学),2014,42(12):1833-1837.
[22] Chen X G,Kong L,Sheng L H,Li X,Zou H F.Chin.J.Anal.Chem.(陈学国,孔亮,盛亮洪,厉欣,邹汉法.分析化学),2005,23(1):46-51.
[23] Lee H,Lim H,Yang J,Hong J.BullKoreanChem.Soc.,2013,34(12):3629-3634.
猜你喜欢
儿童时代·快乐苗苗(2022年10期)2022-12-09
化工管理(2022年14期)2022-12-02
中国化肥信息(2022年3期)2022-05-05
时代邮刊·下半月(2021年10期)2021-10-23
石油石化绿色低碳(2019年6期)2019-02-13
现代园艺(2018年2期)2018-03-15
人民周刊(2016年11期)2016-06-30
中国化肥信息(2016年27期)2016-05-17
中国猪业(2016年1期)2016-01-28
红蜻蜓·低年级(2014年7期)2014-10-20
