
Pulmonary Arterial Hypertension Medical Management of the Adult Patient with Congenital Heart Disease
2018-07-04 06:39AliAtayaMDJulianChungMDJessicaCopePharmDandHassanAlnuaimatMDUniversityofFloridaDivisionofPulmonaryCriticalCareandSleepMedicine600SWArcherRdM45POBox005GainesvilleFLUSA
Ali Ataya, MD, Julian Chung, MD, Jessica Cope, PharmD and Hassan Alnuaimat, MDUniversity of Florida, Division of Pulmonary, Critical Care and Sleep Medicine, 600 SW Archer Rd, M45, PO Box 005,Gainesville, FL, USA
2University of Florida, Department of Pharmacy, Gainesville, FL, USA
lntroduction
Pulmonary arterial hypertension (PAH) is defined by a mean pulmonary arterial pressure of 25 mmHg or greater at rest with a pulmonary capillary wedge pressure of 15 mmHg or less and a pulmonary vascular resistance (PVR) of more than 3 Wood units(WU) [1].Patients with congenital heart diseases(CHDs) represent a heterogeneous patient population because of the differences in underlying cardiac defects.CHD-associated PAH is a well-recognized entity that may develop during infancy through adulthood.Secondary to this, CHD-associated PAH has important prognostic implications because of the associated increased risk of morbidity and two-fold increase in mortality [2].Recent advancements in medical management of PAH in the past 2 decades include the development of PAH-specific therapies that have been associated with improved outcomes;however, limited evidence exists supporting the role of PAH-specific therapies in patients with CHD-associated PAH.
Epidemiology, Pathophysiology, and Classification
With more than 90% of children with CHD reaching childhood, there is an increased prevalence of adult patients with CHD [3, 4].It is estimated that with the growing CHD population, approximately 5–10% of adult patients with CHD will develop PAH [5–7].Pulmonary hypertension may occur in patients with CHD because of increased pulmonary blood fl ow in the setting of systemic-to-pulmonary shunt, increased PVR, or passive venous congestion,or as the result of the combination of these conditions.The end result is a pulmonary arteriopathy and increased PVR similar to that seen in patients with idiopathic PAH.If left untreated, the increasing PVR will surpass the systemic vascular resistance, resulting in reversal of shunt fl ow (right to left) and the development of Eisenmenger syndrome [8].
The most up-to-date classification of pulmonary hypertension categorizes CHD-associated PAH as a subgroup of group 1 PAH [9].The severity of PAH is dependent on the cause of CHD; for example, a higher number of patients with unrepaired ventricular septal defects (VSDs) will end up developing Eisenmenger syndrome and eventually PAH compared with patients with unrepaired atrial septal defects (ASDs) [10].The most recent Nice symposium (2013) classified CHD-associated PAH under four separate phenotypes: (1) Eisenmenger syndrome; (2) left-to-right shunts (correctable and uncorrectable); (3) PAH with coincidental small cardiac defects (ASD <2 cm and VSD <1 cm) that themselves do not account for the development of PAH; (4) PAH that persists or develops after surgical correction of CHD [9].Of the four phenotypes,patients with postoperative CHD-associated PAH have the worst prognosis, with survival rates similar to those seen in idiopathic PAH [11, 12].
Diagnosis
Patients often present with progressive dyspnea with exertion, increased fatigue, lower extremity swelling, and in some cases cyanosis [13].Echocardiogram is often performed initially in the evaluation of pulmonary hypertension to help define the underlying anatomical abnormalities and estimate the right ventricular systolic pressures.An echocardiogram may also identify other markers of elevated pulmonary pressures, such as dilatation of the right cardiac chambers and right ventricular systolic dysfunction.In some cases, cardiac magnetic resonance imaging is performed to more clearly delineate the cardiac defects and provide further measures of right-sided heart function.A diagnosis of PAH can be made only by right-sided heart catheterization in which precapillary pulmonary hemodynamics are observed [1].
Treatment of CHD-Associated PAH
Patients with CHD-associated PAH should be referred to and treated at tertiary centers experienced in treating this unique subpopulation [14].The literature has shown that adult patients with CHD-associated PAH have improved survival and outcomes if they are treated at tertiary centers and are more likely to receive PAH-specific therapies[15, 16].Treatment with PAH-specific therapies in patients with Eisenmenger syndrome has been independently associated with improved survival [17].PAH-specific pharmacotherapy is targeted at the three pathophysiologic pathways: the nitric oxide pathway, the endothelin pathway, and the prostacyclin pathway.The CHD-associated PAH population is largely underrepresented within PAH pharmacotherapy clinical trials, thus making it difficult to extrapolate general PAH results to all patients with CHD-associated PAH.That being said, several small, observational trials support the use PAH-specific therapies among patients with Eisenmenger syndrome and other causes of CHD-associated PAH[18, 19].
Pharmacotherapy targeted at the nitric oxide pathway includes the phosphodiesterase 5 inhibitors sildenafil and tadalafil, as well as the soluble guanylate cyclase stimulator riociguat.In the Sildenafil Use in Pulmonary Arterial Hypertension(SUPER-1) trial, CHD-associated PAH patients constituted 6% of the study population.Overall,patients treated with sildenafil experienced a reduction in functional class, increase in exercise capacity, and improvement in hemodynamics compared with patients who received placebo but data with regard to CHD-associated PAH were not available [20].Additional literature supports the use of sildena fi l in CHD-associated PAH, with favorable effects on functional class, exercise capacity,and hemodynamics (Table 1) [21–24].Similar favorable results were seen in response to tadalafil [25, 26].In Pulmonary Arterial Hypertension Soluble Guanylate Cyclase–Stimulator Trial 1(PATENT-1), patients with CHD-associated PAH with closed defects constituted 8% (n= 35) of the population studied.These patients experienced reduction in functional class, increase in exercise capacity, increase in time to clinical worsening, and improvement in hemodynamic parameters [27].Similarly to the SUPER-1 trial, CHD-speci fi c outcomes were not available, but there was an overall improvement.
Pharmacotherapy targeted at the endothelin pathway includes endothelin receptor antagonists such as bosentan, ambrisentan, and macitentan.Bosentan use in Eisenmenger syndrome resulted in favorable response with regard to exercise capacity and hemodynamics in the Bosentan Randomized Trial of Endothelin Antagonist Therapy-5(BREATHE-5) (Table 2).The response to treatment does not depend on the location of the defect [28,32].In the treatment arm, 65% of patients (n= 24)had VSDs, 22% (n= 8) had ASDs, and 14% (n= 5)had both VSDs and ASDs.Other studies also confirmed favorable responses to bosentan (Table 2)[29–32].Ambrisentan has been associated with increase in the 6-minute walk test distance in patients with CHD-associated PAH at 6 months[33].The Study with an Endothelin Receptor Antagonist in Pulmonary Arterial Hypertension to Improve Clinical Outcome (SERAPHIN) trial included 8.4% of patients (n= 62) with CHD-associated PAH.The study investigators observed reduced morbidity and mortality with treatment with macitentan (Table 3) [34].Additional data on the use of macitentan in Eisenmenger syndrome have been obtained, and their publication is awaited (NCT01743001).Blok et al.[35] observed reduction in functional class and improvement in echocardiogram findings, including right ventricle parameters, in patients with CHD-associated PAH whose treatment was switched from bosentan to macitentan; however, there was no difference in the incidence of syncope or heart failure–associated hospitalizations.
Pharmacotherapy targeted at the prostacyclin pathway includes medications such as epoprostenol,treprostinil, and iloprost, which can be delivered via several routes, including intravenous, subcutaneous, oral, and inhaled routes.The use of intravenous therapy is associated with an increased risk of bloodstream infection and paradoxical embolism, while the subcutaneous and inhaled therapies are associated with injection site pain and frequent dosing,respectively.More recently, an oral form of treprostinil received FDA approval as did an oral prostacyclin receptor agonist, selexipag.Intravenous administration of epoprostenol in CHD-associated PAH has shown beneficial effects on functional class, exercise capacity, and pulmonary hemodynamics (Table 4) [36–38].In a randomized trial of subcutaneously administered treprostinil versus placebo among 109 (23%) patients with CHD-associated PAH, an increase in exercise capacity,particularly in severely ill, was observed.Patients who walked less than 150 m at the baseline on the 6-minute walk test had an increase of 51 ± 16 m(P = 0.002) [39].Inhaled iloprost has been associated with an increase in exercise capacity in patients with Eisenmenger syndrome; however, no change in pulmonary hemodynamics was observed [40].In the Prostacyclin Receptor Agonist in Pulmonary Arterial Hypertension Study (GRIPHON) trial,selexipag was associated with a lower incidence of death and PAH-associated complications,including in patients in the CHD-associated PAH subpopulation [41].
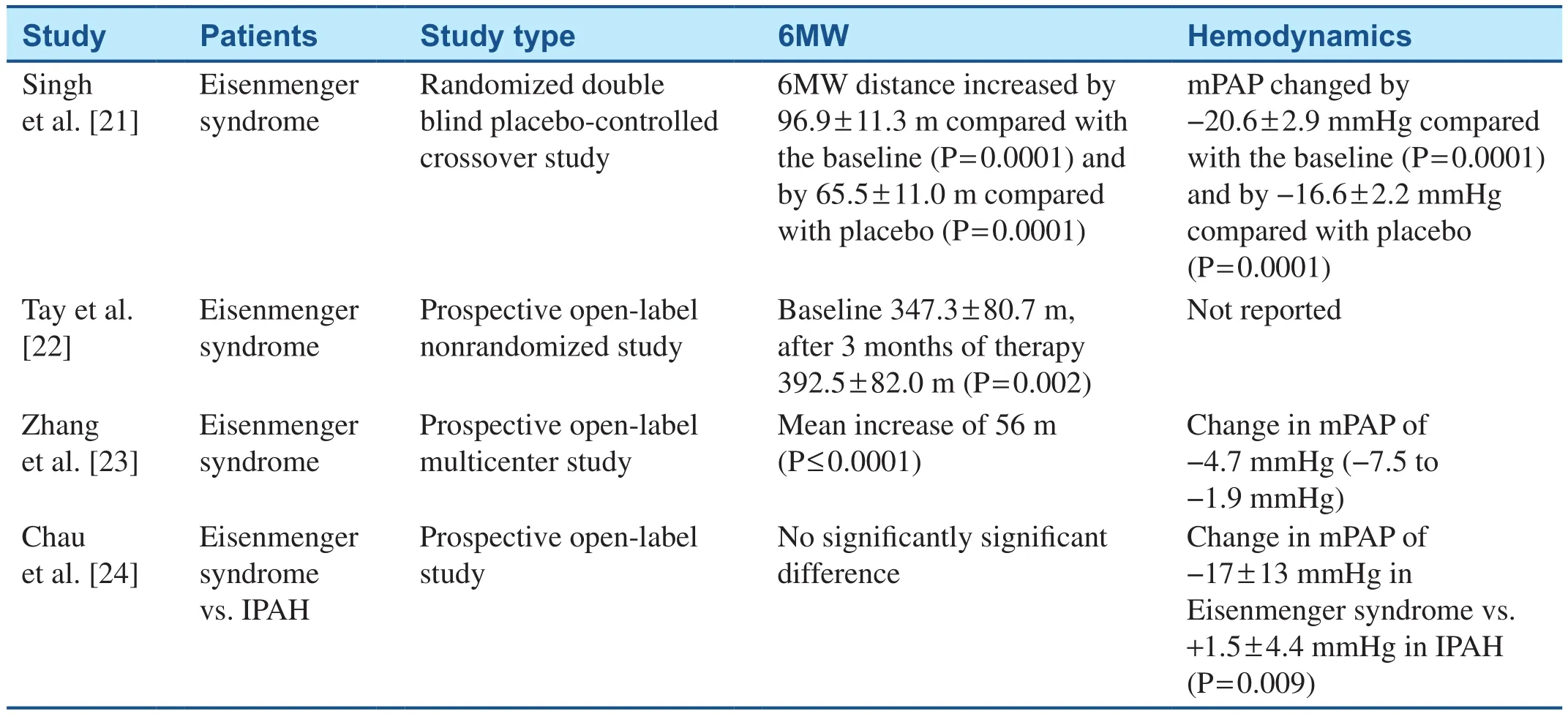
Table 1 Studies Evaluating the Effects of Sildenafil on Patients with Congenital Heart Disease–Associated Pulmonary Arterial Hypertension.
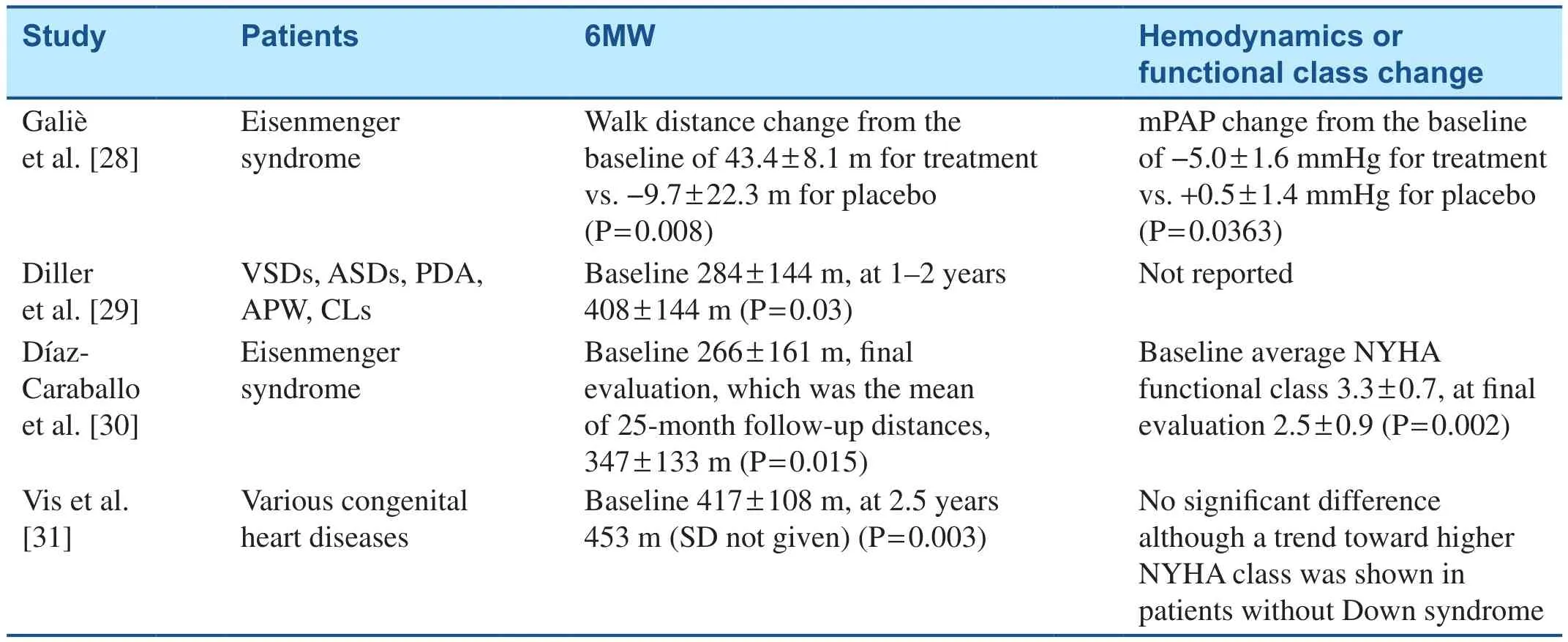
Table 2 Studies Evaluating the Effects of Bosentan on Patients with Congenital Heart Disease–Associated Pulmonary Arterial Hypertension.

Table 3 SERAPHIN Study Evaluating the Effects of Macitentan on Patients with Congenital Heart Disease–Associated Pulmonary Arterial Hypertension [34].
Upfront combination and sequential combination therapy has been shown to be associated with a survival advantage in patients with PAH[42].Sildenafil add-on therapy in patients withCHD-associated PAH receiving bosentan monotherapy has been associated with an increase in exercise capacity and improvement in clinical status and hemodynamics [43].After 6 months of therapy, there was a reduction in World Health Organization functional class (2.1 ± 0.4 vs.2.9 ± 0.3; P = 0.042), an increase in 6-minute walk distance (360 ± 51 m vs.293 ± 68 m, P = 0.005),an improvement in hemodynamics (pulmonary blood fl ow 3.4 ± 1.0 L/min/m2vs.3.1 ± 1.2 L/min/m2, P = 0.002), and a reduction in PVR(19 ± 9 WU/m2vs.24 ± 16 WU/m2, P = 0.003).However, Iverson et al.[44] failed to show the same beneficial effects in patients with CHD-associated PAH with Eisenmenger syndrome receiving sildenafil and bosentan combination therapy.The Ambrisentan and Tadalafil Combination Therapy in Subjects with Pulmonary Arterial Hypertension(AMBITION) study concluded that upfront tadalafil and ambrisentan combination therapy was associated with a lower risk of clinical-failure events compared with monotherapy [45].Patients with CHD-associated PAH composed 2% of the AMBITION study population (n= 13).Clinical trials pursuing upfront triple oral therapy are currently under way in patients with PAH, including the Efficacy and Safety of Initial Triple versus Initial Dual Oral Combination Therapy in Patients with Newly Diagnosed Pulmonary Arterial Hypertension (TRITON) trial (NCT02558231).Further studies are under way addressing different combination therapies in PAH patients, including those with CHD-associated PAH.

Table 4 Studies Evaluating the Effects of Epoprostenol on Patients with Congenital Heart Disease–Associated Pulmonary Arterial Hypertension.
Treat-to-Close Strategy
The treat-to-close strategy was investigated specifically in patients with ASDs and PAH.Among 22 patients undergoing closure of their ASD, the investigators found that use of PAH-specific therapy before closure for 36% of patients (n= 8) resulted in significant symptomatic relief before transcatheter shunt closure without any procedure-related complications or adverse events [46].Before treatment and closure the group treated with PAH-specific therapies had significantly worse hemodynamics on the basis of a higher mean pulmonary arterial pressure (62 ± 21 mmHg vs.35 ± 8 mmHg, P ≤ 0.01) and lower PVR (9.6 ± 3.8 WU vs.41 ± 1.1 WU, P ≤ 0.01).After starting medical therapy, the two groups had similar hemodynamic profiles and were successfully treated with transcatheter closure.This study may indicate that treatment as a bridge to closure may be successful in other types of CHD patients, but further research to evaluate this approach is needed.
Conclusion
In patients with CHD-associated PAH, PAH-specific therapy appears to be an effective method in increasing functional capacity, reducing functional class,and producing more favorable pulmonary vascular hemodynamics.Multiple modalities of therapies appear to be effective in treatment among patients with unrepaired as well as repaired defects in a variety of cardiac abnormalities.Although the data are limited, the growing body of evidence seems to support pharmacotherapy in this heterogeneous patient population as the sole available therapy, and in the future it may prove useful as treatment in preparation for surgical intervention.
Conflict of lnterest
The authors declare that they have no conflicts of interest
Author Contributions
All authors contributed to the content and the writing of the article.Hassan Alnuaimat takes full responsibility for the content of the article.
REFERENCES
1.Hoeper MM, Bogaard HJ, Condliffe R, Frantz R, Khanna D, Kurzyna M,et al.Definitions and diagnosis of pulmonary hypertension.J Am Coll Cardiol 2013;62(25):D42–50.
2.Lowe BS, Therrien J, Ionescu-Ittu R, Pilote L, Martucci G, Marelli AJ.Diagnosis of pulmonary hypertension in the congenital heart disease adult population impact on outcomes.J Am Coll Cardiol 2011;58(5):538–46.
3.Moons P, Bovijn L, Budts W,Belmans A, Gewillig M.Temporal trends in survival to adulthood among patients born with congenital heart disease from 1970 to 1992 in Belgium.Circulation 2010;122(22):2264–72.
4.Marelli AJ, Mackie AS, Ionescu-Ittu R, Rahme E, Pilote L.Congenital heart disease in the general population: changing prevalence and age distribution.Circulation 2007;115(2):163–72.
5.Duffels MG, Engelfriet PM, Berger RM, van Loon RL, Hoendermis E,Vriend JW, et al.Pulmonary arterial hypertension in congenital heart disease: an epidemiologic perspective from a Dutch registry.Int J Cardiol 2007;120(2):198–204.
6.van Riel AC, Schuuring MJ, van Hessen ID, Zwinderman AH,Cozijnsen L, Reichert CL, et al.Contemporary prevalence of pulmonary arterial hypertension in adult congenital heart disease following the updated clinical classification.Int J Cardiol 2014;174(2):299–305.
7.Engelfriet PM, Duffels MG, Moller T, Boersma E, Tijssen JG, Thaulow E, et al.Pulmonary arterial hypertension in adults born with a heart septal defect: the Euro Heart Survey on adult congenital heart disease.Heart 2007;93(6):682–7.
8.Beghetti M, Galie N.Eisenmenger syndrome a clinical perspective in a new therapeutic era of pulmonary arterial hypertension.J Am Coll Cardiol 2009;53(9):733–40.
9.Simonneau G, Gatzoulis MA,Adatia I, Celermajer D, Denton C,Ghofrani A, et al.Updated clinical classification of pulmonary hypertension.J Am Coll Cardiol 2013;62(25 Suppl):D34–41.
10.Van de Bruaene A, Delcroix M,Pasquet A, De Backer J, De Pauw M, Naeije R, et al.The Belgian Eisenmenger syndrome registry:implications for treatment strategies?Acta Cardiol 2009;64(4):447–53.
11.Alonso-Gonzalez R, Lopez-Guarch CJ, Subirana-Domenech MT, Ruiz JM, Gonzalez IO,Cubero JS, et al.Pulmonary hypertension and congenital heart disease: an insight from the REHAP National Registry.Int J Cardiol 2015;184:717–23.
12.Manes A, Palazzini M, Leci E,Bacchi Reggiani ML, Branzi A,Galie N.Current era survival of patients with pulmonary arterial hypertension associated with congenital heart disease: a comparison between clinical subgroups.Eur Heart J 2014;35(11):716–24.
13.Diller GP, Dimopoulos K, Okonko D, Li W, Babu-Narayan SV, Broberg CS, et al.Exercise intolerance in adult congenital heart disease: comparative severity, correlates, and prognostic implication.Circulation 2005;112(6):828–35.
14.Galie N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A,et al.2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society(ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT).Eur Heart J 2016;37(1):67–119.
15.Mylotte D, Pilote L, Ionescu-Ittu R, Abrahamowicz M, Khairy P,Therrien J, et al.Specialized adult congenital heart disease care: the impact of policy on mortality.Circulation 2014;129(18):1804–12.
16.Diller GP, Korten MA, Bauer UM,Miera O, Tutarel O, Kaemmerer H,et al.Current therapy and outcome of Eisenmenger syndrome: data of the German National Register for congenital heart defects.Eur Heart J 2016;37(18):1449–55.
17.Arnott C, Strange G, Bullock A,Kirby AC, O’Donnell C, Radford DJ, et al.Pulmonary vasodilator therapy is associated with greater survival in Eisenmenger syndrome.Heart 2017.doi:10.1136/heartjnl-2017-311876.
18.Dimopoulos K, Inuzuka R, Goletto S, Giannakoulas G, Swan L, Wort SJ, et al.Improved survival among patients with Eisenmenger syndrome receiving advanced therapy for pulmonary arterial hypertension.Circulation 2010;121(1):20–5.
19.Adriaenssens T, Delcroix M, Van Deyk K, Budts W.Advanced therapy may delay the need for transplantation in patients with the Eisenmenger syndrome.Eur Heart J 2006;27(12):1472–7.
20.Galie N, Ghofrani HA, Torbicki A, Barst RJ, Rubin LJ, Badesch D,et al.Sildenafil citrate therapy for pulmonary arterial hypertension.N Engl J Med 2005;353(20):2148–57.
21.Singh TP, Rohit M, Grover A,Malhotra S, Vijayvergiya R.A randomized, placebo-controlled, doubleblind, crossover study to evaluate the efficacy of oral sildenafil therapy in severe pulmonary artery hypertension.Am Heart J 2006;151(4):851.e1–5.
22.Tay EL, Papaphylactou M, Diller GP, Alonso-Gonzalez R, Inuzuka R, Giannakoulas G, et al.Quality of life and functional capacity can be improved in patients with Eisenmenger syndrome with oral sildenafil therapy.Int J Cardiol 2011;149(3):372–6.
23.Zhang ZN, Jiang X, Zhang R, Li XL, Wu BX, Zhao QH, et al.Oral sildenafil treatment for Eisenmenger syndrome: a prospective, openlabel, multicentre study.Heart 2011;97(22):1876–81.
24.Chau EM, Fan KY, Chow WH.Effects of chronic sildenafil in patients with Eisenmenger syndrome versus idiopathic pulmonary arterial hypertension.Int J Cardiol 2007;120(3):301–5.
25.Mukhopadhyay S, Nathani S, Yusuf J, Shrimal D, Tyagi S.Clinical efficacy of phosphodiesterase-5 inhibitor tadalafil in Eisenmenger syndrome – a randomized, placebocontrolled, double-blind crossover study.Congenit Heart Dis 2011;6(5):424–31.
26.Mukhopadhyay S, Sharma M,Ramakrishnan S, Yusuf J, Gupta MD,Bhamri N, et al.Phosphodiesterase-5 inhibitor in Eisenmenger syndrome:a preliminary observational study.Circulation 2006;114(17):1807–10.
27.Ghofrani HA, Galie N, Grimminger F, Grunig E, Humbert M, Jing ZC,et al.Riociguat for the treatment of pulmonary arterial hypertension.N Engl J Med 2013;369(4):330–40.
28.Galiè N, Beghetti M, Gatzoulis MA,Granton J, Berger RM, Lauer A,et al.Bosentan therapy in patients with Eisenmenger syndrome: a multicenter, double-blind, randomized, placebo-controlled study.Circulation 2006;114(1):48–54.
29.Diller GP, Dimopoulos K, Kaya MG, Harries C, Uebing A, Li W, et al.Long-term safety, tolerability and efficacy of bosentan in adults with pulmonary arterial hypertension associated with congenital heart disease.Heart 2007;93(8):974–6.
30.Díaz-Caraballo E, González-García AE, Reñones M, Sánchez-Recalde A, García-Río F, Oliver-Ruiz JM.Long-term bosentan treatment of complex congenital heart disease and Eisenmenger’s syndrome.Rev Esp Cardiol 2009;62(9):1046–9.
31.Vis JC, Duffels MG, Mulder P,de Bruin-Bon RH, Bouma BJ,Berger RM, et al.Prolonged beneficial effect of bosentan treatment and 4-year survival rates in adult patients with pulmonary arterial hypertension associated with congenital heart disease.Int J Cardiol 2013;164(1):64–9.
32.Berger RM, Beghetti M, Galie N,Gatzoulis MA, Granton J, Lauer A, et al.Atrial septal defects versus ventricular septal defects in BREATHE-5, a placebo-controlled study of pulmonary arterial hypertension related to Eisenmenger’s syndrome: a subgroup analysis.Int J Cardiol 2010;144(3):373–8.
33.Zuckerman WA, Leaderer D, Rowan CA, Mituniewicz JD, Rosenzweig EB.Ambrisentan for pulmonary arterial hypertension due to congenital heart disease.Am J Cardiol 2011;107(9):1381–5.
34.Pulido T, Adzerikho I, Channick RN, Delcroix M, Galie N, Ghofrani HA, et al.Macitentan and morbidity and mortality in pulmonary arterial hypertension.N Engl J Med 2013;369(9):809–18.
35.Blok IM, van Riel A, van Dijk APJ,Mulder BJM, Bouma BJ.From bosentan to macitentan for pulmonary arterial hypertension and adult congenital heart disease: further improvement? Int J Cardiol 2017;227:51–2.
36.Fernandes SM, Newburger JW,Lang P, Pearson DD, Feinstein JA, Gauvreau K, et al.Usefulness of epoprostenol therapy in the severely ill adolescent/adult with Eisenmenger physiology.Am J Cardiol 2003;91(5):632–5.
37.Rosenzweig EB, Kerstein D, Barst RJ.Long-term prostacyclin for pulmonary hypertension with associated congenital heart defects.Circulation 1999;99(14):1858–65.
38.Thomas IC, Glassner-Kolmin C,Gomberg-Maitland M.Long-term effects of continuous prostacyclin therapy in adults with pulmonary hypertension associated with congenital heart disease.Int J Cardiol 2013;168(4):4117–21.
39.Simonneau G, Barst RJ, Galie N,Naeije R, Rich S, Bourge RC, et al.Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a doubleblind, randomized, placebo-controlled trial.Am J Respir Crit Care Med 2002;165(6):800–4.
40.Cha KS, Cho KI, Seo JS, Choi JH,Park YH, Yang DH, et al.Effects of inhaled iloprost on exercise capacity, quality of life, and cardiac function in patients with pulmonary arterial hypertension secondary to congenital heart disease (the Eisenmenger syndrome) (from the EIGER study).Am J Cardiol 2013;112(11):1834–9.
41.Sitbon O, Channick R, Chin KM,Frey A, Gaine S, Galie N, et al.Selexipag for the treatment of pulmonary arterial hypertension.N Engl J Med 2015;373(26):2522–33.
42.Fox BD, Shtraichman O, Langleben D, Shimony A, Kramer MR.Combination therapy for pulmonary arterial hypertension: a systematic review and meta-analysis.Can J Cardiol 2016;32(12):1520–30.
43.D’Alto M, Romeo E, Argiento P,Sarubbi B, Santoro G, Grimaldi N,et al.Bosentan-sildenafil association in patients with congenital heart disease-related pulmonary arterial hypertension and Eisenmenger physiology.Int J Cardiol 2012;155(3):378–82.
44.Iversen K, Jensen AS, Jensen TV,Vejlstrup NG, Sondergaard L.Combination therapy with bosentan and sildenafil in Eisenmenger syndrome: a randomized, placebocontrolled, double-blinded trial.Eur Heart J 2010;31(9):1124–31.
45.Galie N, Barbera JA, Frost AE,Ghofrani HA, Hoeper MM,McLaughlin VV, et al.Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension.N Engl J Med 2015;373(9):834–44.
46.Kijima Y, Akagit, Takaya Y,Akagi S, Nakagawa K, Kusano K, et al.Treat and repair strategy in patients with atrial septal defect and significant pulmonary arterial hypertension.Circ J 2016;80(1):227–34.
 Cardiovascular Innovations and Applications2018年2期
Cardiovascular Innovations and Applications2018年2期
- Cardiovascular Innovations and Applications的其它文章
- The Pulmonary Hypertension Story
- The Surgical Management of Ebstein Anomaly
- Evaluation of Left Ventricular Systolic Function after Pulmonary Valve Replacement Using Cardiovascular Magnetic Resonance lmaging
- Pregnancy in Congenital Heart Disease:A Review for the General Cardiologist
- Atrial Arrhythmias lncluding Atrial Fibrillation in Congenital Heart Disease: Mechanisms,Substrate ldentification and lnterventional Approaches
- Heart Transplantation for Adult Congenital Heart Disease: Overview and Special Considerations