
芍药苷减轻糖尿病小鼠肾组织炎症与JAK2/STAT3信号通路的关系
2018-07-04 11:02李新玉邵云侠吴永贵
安徽医科大学学报 2018年7期
李新玉,邵云侠,王 坤,吴永贵
糖尿病肾脏疾病(diabetic kidney disease, DKD)是导致终末期肾脏疾病(end stage renal disease,ESRD)的主要原因之一[1]。研究[2]表明肾脏炎症在DKD发病机制中发挥着重要作用。巨噬细胞是炎症反应的主要调节细胞,巨噬细胞的浸润和极化介导的肾脏损伤与DKD白蛋白尿、肾脏纤维化及肾功能进行性下降密切相关[3]。酪氨酸蛋白激酶/信号转导与转录激活子(janus activated kinase/signal transducer and activator of transcriptions, JAK /STAT)信号通路是介导肾脏炎症反应的一条主要信号通路,其中的JAK2/STAT3亚型是目前肾脏疾病中研究最广泛和深入的一种[4]。高糖环境、血管紧张素Ⅱ、晚期糖基化终产物、过氧化应激均能激活JAK2/STAT3通路,从而引起DKD肾组织炎症[4]。芍药苷(paeoniflorin, PF)是中药白芍总苷中的主要生物活性物质,具有抗炎、免疫调节、抗氧化等药理学作用[5]。先前的研究[6]证明PF可抑制DKD动物模型的肾脏炎症反应。该研究主要从JAK2/STAT3信号通路方面探讨PF对糖尿病小鼠肾损伤的影响及可能机制。
1 材料与方法
1.1材料与动物
1.1.1动物 雄性健康SPF级C57BL/6J小鼠60只,8~10周龄,18~20 g,由安徽医科大学动物中心提供。所有小鼠在整个实验期间给予标准饮食,自由饮水,适应性饲养1周,观察12周。
1.1.2药品和试剂 PF(南京广润生物制品有限公司);链脲佐菌素(streptozocin, STZ)试剂(美国 Sigma公司);尿白蛋白测定试剂盒(美国Abcam公司);通用二步法检测试剂盒(PV-9000)、DAB显色液(北京中杉金桥生物技术有限公司);鼠抗CD68多克隆抗体(美国Santa Cruz公司);兔抗JAK2、兔抗STAT3、兔抗p-JAK2、兔抗p-STAT3多克隆抗体(美国Cell Signaling公司);小鼠抗β-actin单克隆抗体、辣根过氧化物酶标记羊抗兔/鼠IgG(武汉三鹰生物技术有限公司);BCA蛋白定量试剂盒(碧云天生物工程研究所);ECL发光试剂盒(美国Thermo Scientific公司);硝酸纤维(nitrocellulose membrane,NC)膜(美国Millipore公司);TRIzol试剂(美国Life Technologie公司);反转录试剂盒(美国Promega公司);实时定量PCR试剂盒(美国Bio-Rad公司);实时定量PCR引物(上海生工生物工程股份有限公司、美国Gene Copoeia公司)。
1.2方法
1.2.1动物分组 适应性饲养1周后的小鼠随机分为5组:对照组、模型组、PF 25 mg/kg组、PF 50 mg/kg组、PF 100 mg/kg组,每组12只。除对照组外,其余各组小鼠连续5 d腹腔注射50 mg/kg STZ,对照组注射同等剂量枸橼酸缓冲液。1周后小鼠尾静脉采血,测定随机血糖为16.7 mmol/L以上提示糖尿病模型造模成功,纳入后续实验。PF每天分别以25、50、100 mg/kg的剂量腹腔注射给药,对照组和模型组给予等量溶媒。
1.2.2标本收集 实验12周末将小鼠置于代谢笼中收集24 h尿液标本,离心后分装好置于-80 ℃冰箱中冻存,用于测定24 h尿白蛋白含量。小鼠腹腔注射麻醉后记录体质量,眶静脉采血,离心后用于测血糖水平。切取右侧肾脏,去除被膜及肾周脂肪记录肾重,切割成小块置于液氮罐中冻存,用于Western blot及实时定量PCR检测;左侧肾脏剥离被膜后置于4%多聚甲醛中固定,用于病理组织学观察及免疫组化检测。
1.2.3尿白蛋白排泄率(urinary albumin excretion rate, UAER)测定 取小鼠尿液标本离心,使用小鼠尿白蛋白试剂盒通过ELISA法测定各组小鼠尿白蛋白含量。小鼠UAER为尿白蛋白含量与24 h 尿量乘积。
1.2.4肾组织病理学检测 肾组织标本经脱水、石蜡包埋制成组织切片后行肾组织PAS染色。每组标本随机选取10个高倍视野(×400)下的肾小球和肾小管间质区进行肾组织病理学评分。肾小球系膜增生指数评分:根据系膜增生面积,按正常、轻度(<25%)、中度(25%~50%)、中重度(50%~75%)、重度(>75%),分别计0、1、2、3、4分。肾小管间质损伤指数评分:根据肾小管萎缩与扩张、间质炎症与纤维化严重程度,按正常、轻度(面积<25%)、中度(面积达25%~50%)、重度(面积在>50%),分别计0、1、2、3分。计算每片切片肾小球及肾小管间质组织学评分的平均值。
1.2.5肾组织免疫组化检测 小鼠肾组织石蜡切片脱蜡后,经3% H2O2封闭内源性过氧化酶、微波炉修复表面抗原、山羊血清封闭,分别滴加适量一抗:CD68抗体(1 ∶50)、p-JAK2抗体(1 ∶100)及p-STAT3抗体(1 ∶100),4 ℃过夜,复温后加入二抗,二抗使用通用二步法检测试剂:先滴加PV-9000试剂1,37 ℃孵育20 min后再滴加PV-9000试剂2,37 ℃孵育30 min后滴加 DAB 显色液显色。每组标本随机选取10个高倍视野(×400)下的肾小球及肾小管间质区域,通过Image-Pro-Plus 6.0分析系统分别计算肾组织CD68免疫组化切片肾小球及肾小管间质区阳性细胞个数及肾组织p-JAK2、p-STAT3免疫组化切片肾小球和肾小管间质着色阳性面积百分比(%),取均值进行比较。
1.2.6肾组织Western blot检测 小鼠肾组织裂解后提取总蛋白,取蛋白样品煮沸后经SDS-PAGE电泳后电转移至NC膜上,洗膜后置于5%脱脂奶粉-TBST溶液中,37 ℃封闭2 h,洗膜后放入一抗中: p-JAK2抗体(1 ∶1 000)、JAK2抗体(1 ∶1 000)、p-STAT3抗体(1 ∶1 000)、STAT3抗体(1 ∶1 000)、β-actin抗体(1 ∶35 000),4 ℃孵育过夜,洗膜后再加入二抗:辣根过氧化物酶标记羊抗兔/鼠IgG(1 ∶35 000),37 ℃孵育1 h。洗膜后置于ECL发光试剂中显色,经成像系统曝光显影。以管家基因β-actin作为内参,通过Image J软件计算Western blot条带光密度(optical density,OD)值,结果以p-JAK2/JAK2、p-STAT3/STAT3比值的均值为蛋白表达的相对量。
1.2.7肾组织实时定量PCR检测 各组小鼠肾组织总RNA通过TRIzol试剂提取,分光光度计测定各组OD值,以 OD260 nm/OD280 nm比值在1.8~2.0之间为样本RNA纯净。再以RNA为模板逆转录成cDNA。采用SYBR Green试剂盒进行PCR,每组样本重复检测3次,样本基因的相对表达量通过2-ΔΔCt方式计算。GAPDH引物序列上游引物:5′-GGTGAAGGTCGGTGTGAACG-3′;下游引物:5′-CTCGCTCCTGGAAGATGGTG-3′。肿瘤坏死因子α(tumor necrotic factor-α, TNF-α)引物序列上游引物:5′-GCTGAGCTCAAACCCTGGTA-3′;下游引物:5′-CGGACTCCG CAAAGTCTAAG-3′。以下引物由Gene Copoeia公司合成:白细胞介素1β(interleukin-1β, IL-1β)货号MQP027422;单核细胞趋化因子-1(monocyte chemoattractant protein-1, MCP-1)货号MQP027672;诱生型一氧化氮合酶(inducible nitric oxide synthase, iNOS)货号MQP029793。

2 结果
2.1各组小鼠一般情况变化对照组小鼠精神良好、进食饮水正常、反应迅速、毛色光亮,模型组小鼠逐渐出现精神萎靡、多饮多食、尿量增加、反应迟钝、毛色污秽等糖尿病症状,PF 25、50、100 mg/kg给药组上述表现较模型组明显减轻。
2.2各组小鼠一般指标变化模型组小鼠血糖明显高于对照组(P<0.01),而PF 25、50、100 mg/kg给药组与模型组小鼠血糖水平差异无统计学意义(P>0.05),且仍明显高于对照组(P<0.01),表明PF给药对血糖水平没有影响。模型组小鼠的相对肾重明显高于对照组(P<0.05),与模型组相比,PF 25、50、100 mg/kg给药12周后,相对肾重均明显减轻(P<0.05),但仍高于对照组。模型组小鼠UAER明显高于对照组(P<0.01),PF 25、50、100 mg/kg给药12周,小鼠UAER水平较模型组明显减低(P<0.01),且呈剂量依赖性,但仍高于对照组。血糖、相对肾重、UAER的各组间差异有统计学意义(F=74.87,P<0.01;F=3.13,P<0.05;F=351.73,P<0.01)。见表1。
2.3各组小鼠肾组织病理学改变PAS染色结果提示模型组肾小球系膜增生指数和肾小管间质损伤指数评分明显高于对照组(P<0.05),表明模型组小鼠出现DKD早期损伤。PF 25、50、100 mg/kg给药组肾小球系膜增生指数评分和肾小管间质损伤指数评分则均明显低于模型组(P<0.05),表明PF给药后肾脏组织学损伤显著减轻。肾小球系膜增生指数和肾小管间质损伤指数的各组间差异有统计学意义(F=36.60、44.08,P<0.01)。见图1、表2。
2.4各组小鼠肾组织CD68表达本实验选择巨噬细胞表面蛋白CD68标记肾组织巨噬细胞浸润。免疫组化结果显示,对照组肾小球与肾小管间质CD68阳性巨噬细胞浸润数目较少,而模型组肾小球及肾小管-间质可见大量CD68阳性巨噬细胞浸润,明显高于对照组(P<0.01)。与模型组相比,PF 25、50、100 mg/kg给药组肾小球及肾小管-间质CD68阳性巨噬细胞浸润数量则明显减少(P<0.05,P<0.01),各组间差异有统计学意义(F=177.29、36.10,P<0.01)。见图2、表3。
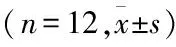
表1 各组小鼠一般指标变化
与对照组比较:*P<0.05,**P<0.01;与模型组比较:#P<0.05,##P<0.01
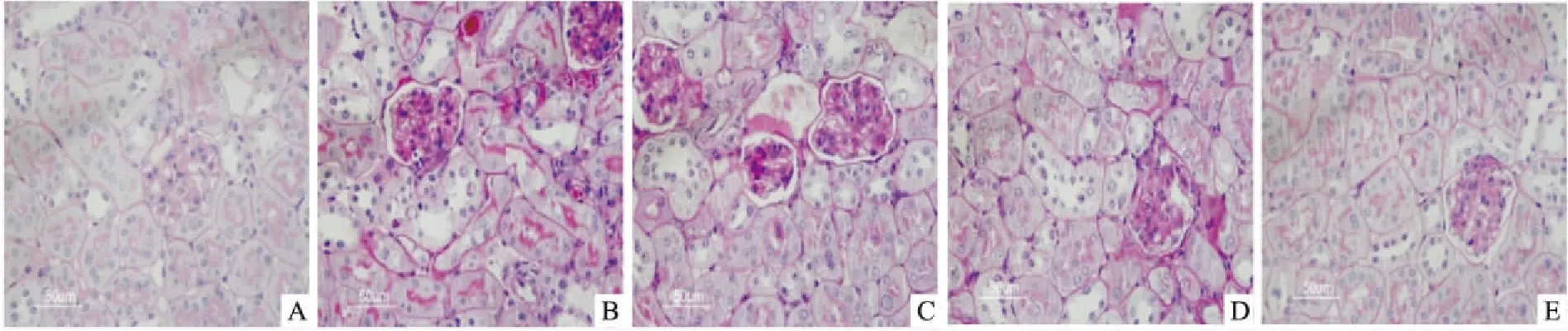
图1 PAS染色肾组织病理形态学 ×400

图2 小鼠肾组织CD68免疫组化 ×400

项目对照组模型组PF 25 mg/ kg组PF 50 mg/ kg组PF 100 mg/ kg组肾小球系膜增生指数0.35±0.102.16±0.59∗1.14±0.38#1.02±0.23#1.17±0.13#肾小管间质损伤指数0.50±0.122.43±0.55∗1.47±0.28#1.29±0.25#1.01±0.34#
与对照组比较:*P<0.05;与模型组比较:#P<0.05
表3 各组小鼠肾组织CD68、p-JAK2与p-STAT3表达变化
与对照组比较:**P<0.01;与模型组比较:#P<0.05;##P<0.01

图3 小鼠肾组织p-JAK2免疫组化 ×400

图4 小鼠肾组织p-STAT3免疫组化 ×400
2.5各组小鼠肾组织p-JAK2与p-STAT3蛋白表达免疫组化结果显示,对照组小鼠肾小球与肾小管-间质p-JAK2与p-STAT3蛋白表达微弱。模型组肾小球与肾小管间质p-JAK2蛋白表达均明显高于对照组(P<0.01),与模型组相比,PF给药明显抑制了肾小球及肾小管间质p-JAK2蛋白表达(P<0.05,P<0.01),各组间差异有统计学意义(F=117.35、91.58,P<0.01)。但模型组仅肾小管间质p-STAT3蛋白表达明显高于对照组(P<0.01),同样,PF给药后,肾小管间质p-STAT3蛋白的高表达也受到明显抑制(P<0.05,P<0.01),各组间差异有统计学意义(F=199.5,P<0.01)。见表3、图3、4。
Western blot杂交条带光密度结果显示,模型组p-JAK2/JAK2与p-STAT3/STAT3蛋白的表达较对照组明显增加(P<0.01)。与模型组相比,PF 25、50、100 mg/kg给药12周则均可使肾组织p-JAK2/JAK2与p-STAT3/STAT3蛋白表达不同程度下降(P<0.01),各组间差异有统计学意义(F=157.78、242.18,P<0.01)。见图5。
图5 Western blot检测肾组织p-JAK2/JAK2与p-STAT3/STAT3蛋白表达
1:对照组;2:模型组;3:PF 25 mg/kg组;4:PF 50 mg/kg组;5:PF 100 mg/kg组;与对照组比较:**P<0.01;与模型组比较:##P<0.01
2.6各组小鼠肾组织TNF-α、IL-1β、MCP-1和iNOSmRNA表达实时定量PCR显示模型组肾组织TNF-α、IL-1β、MCP-1和iNOS mRNA表达较对照组明显升高(P<0.01)。而与模型组相比,PF 25、50、100 mg/kg给药组肾组织TNF-α、IL1-β、MCP-1和iNOS mRNA表达量均不同程度的明显减少(P<0.01)。各指标组间差异有统计学意义(FTNF-α= 460.07、FIL1-β=518.34、FMCP-1=232.64、FiNOS=715.78,P<0.01)。见图6。
3 讨论
DKD作为糖尿病最严重的并发症之一,其主要发病机制与高糖环境介导下细胞内代谢状态的改变有关,炎症作为主要影响因素,近些年来受到越来越多的关注[7]。中药白芍总苷在许多炎症性疾病中发挥着重要的抗炎及免疫调节作用,目前已经被广泛应用于类风湿性关节炎、干燥综合征及系统性红斑狼疮等多种疾病的治疗[8]。PF是白芍总苷的主要生物活性物质,然而目前PF对DKD的具体保护机制尚不明确。微量白蛋白尿是DKD的早期临床表现及诊断依据。本研究结果表明在不依赖于血糖水平减低的情况下,PF给药能够显著减少糖尿病小鼠尿白蛋白排泄率水平,说明PF对糖尿病小鼠肾组织有重要的保护作用。DKD肾脏组织病理特征主要包括肾小球肥大、基底膜增厚、系膜细胞及系膜基质增生等。本研究结果表明PF给药能明显改善糖尿病小鼠肾小球及肾小管间质组织学损伤,在肾脏病理学水平也支持了其肾脏保护作用。

图6 实时定量PCR检测肾组织TNF-α、IL-1β、MCP-1与iNOS mRNA表达
1:对照组;2:模型组;3:PF 25 mg/kg组;4:PF 50 mg/kg组;5:PF 100 mg/kg组;与对照组比较:**P<0.01;与模型组比较:##P<0.01
JAK/STAT通路的激活在DKD发病机制中的作用受到广泛关注。细胞因子或生长激素与其受体结合后,激活与受体偶联的酪氨酸蛋白激酶JAKs,随后募集下游的转录因子STATs磷酸化并转入细胞核内,从而调控靶基因的转录表达[9]。研究[10]表明,JAK/STAT信号通路,尤其是JAK2/ STAT3的活化,上调相关转录因子的表达,导致了高糖环境下肾脏细胞的炎症浸润,细胞增殖以及纤维化。Lu et al[11]通过STAT3基因敲除小鼠模型抑制JAK/STAT通路的激活,观察到了肾组织炎症及损伤减轻作用。同样,JAK/STAT通路激活的负向调节因子细胞因子信号抑制物 (suppressor of cytokine signaling, SOCS),主要包括SOCS1与 SOCS3,两者的过表达均可以减少STAT1及STAT3的激活,从而减轻DKD肾脏损伤[12]。因此,抑制JAK2/STAT3信号通路介导的炎症反应途径可能成为治疗DKD的潜在有效方法。本研究表明糖尿病小鼠肾组织p-JAK2与p-STAT3蛋白表达较对照组明显升高,证实了高糖环境下JAK2/STAT3信号通路的活化,进一步证实PF给药能够显著减少p-JAK2与p-STAT3蛋白表达,提示PF可能通过抑制JAK2/STAT3信号通路激活,减轻肾组织炎症。
巨噬细胞肾脏内的浸润在DKD炎症中至关重要,近年来的研究证明浸润的巨噬细胞表型是最终决定DKD发展结局的重要因素。经典活化型(classically activated,M1型)巨噬细胞促进炎症反应及组织损伤,替代活化型(alternatively activated,M2型)巨噬细胞参与抗炎及组织修复[13]。促炎细胞因子激动M1型巨噬细胞的活化,进一步促进TNF-α、IL-1β、MCP-1等炎症因子的分泌。目前应用最广泛的鉴定M1型巨噬细胞表面分子标志为iNOS[14]。作为调节炎症反应的重要信号通路,JAK/STAT通路的活化能够促进M1型巨噬细胞极化[15]。本实验结果表明,糖尿病模型组小鼠肾组织CD68阳性巨噬细胞浸润数量较对照组显著增多,同时其iNOS的表达显著增加。PF给药后糖尿病小鼠肾组织肾小球及肾小管间质的巨噬细胞浸润则明显减少,并导致iNOS表达显著下降,同时促炎因子TNF-α、IL-1β、MCP-1的表达也明显减少。因此,PF在DKD中的肾脏保护作用与可能抑制巨噬细胞的活化有关,然而在DKD中PF是否通过JAK2/ STAT3信号通路调节M1型巨噬细胞激活发挥其肾脏保护作用及具体机制仍有待进一步的实验探究。
[1] Tuttle K R, Bakris G L, Bilous R W, et al. Diabetic kidney disease: a report from an ADA Consensus Conference[J]. Am J Kidney Dis, 2014,64(4):510-33.
[2] Tesch G H. Diabetic nephropathy - is this an immune disorder?[J]. Clin Sci (Lond), 2017,131(16):2183-99.
[3] Awad A S, You H, Gao T, et al. Macrophage-derived tumor necrosis factor-alpha mediates diabetic renal injury[J]. Kidney Int, 2015,88(4):722-33.
[4] Brosius F C 3rd, He J C. JAK inhibition and progressive kidney disease[J]. Curr Opin Nephrol Hypertens, 2015,24(1):88-95.
[5] Wang C, Yuan J, Wu H X, et al. Paeoniflorin inhibits inflammatory responses in mice with allergic contact dermatitis by regulating the balance between inflammatory and anti-inflammatory cytokines[J]. Inflamm Res, 2013,62(12):1035-44.
[6] Zhang T, Zhu Q, Shao Y, et al. Paeoniflorin prevents TLR2/4-mediated inflammation in type 2 diabetic nephropathy[J]. Biosci Trends, 2017,11(3):308-18.
[7] Turkmen K. Inflammation, oxidative stress, apoptosis, and autophagy in diabetes mellitus and diabetic kidney disease: the four horsemen of the apocalypse[J]. Int Urol Nephrol, 2017,49(5):837-44.
[8] He D Y, Dai S M. Anti-inflammatory and immunomodulatory effects of paeonia lactiflora pall., a traditional chinese herbal medicine[J]. Front Pharmacol, 2011,2:10.
[9] Jenkins B J. Transcriptional regulation of pattern recognition receptors by Jak/STAT signaling, and the implications for disease pathogenesis[J]. J Interferon Cytokine Res, 2014,34(10):750-8.
[10] Brosius F C, Tuttle K R, Kretzler M. JAK inhibition in the treatment of diabetic kidney disease[J]. Diabetologia, 2016,59(8):1624-7.
[11] Lu T C, Wang Z H, Feng X, et al. Knockdown of Stat3 activity in vivo prevents diabetic glomerulopathy[J]. Kidney Int, 2009,76(1):63-71.
[12] Linossi E M, Babon J J, Hilton D J, et al. Suppression of cytokine signaling: the SOCS perspective[J]. Cytokine Growth Factor Rev, 2013,24(3):241-8.
[13] Meng X M, Tang P M, Li J, et al. Macrophage phenotype in kidney injury and repair[J]. Kidney Dis (Basel), 2015,1(2):138-46.
[14] Guiteras R, Flaquer M, Cruzado J M. Macrophage in chronic kidney disease[J]. Clin Kidney J, 2016,9(6):765-71.
[15] Zhou D, Huang C, Lin Z, et al. Macrophage polarization and function with emphasis on the evolving roles of coordinated regulation of cellular signaling pathways[J]. Cell Signal, 2014,26(2):192-7.
猜你喜欢
临床与实验病理学杂志(2021年7期)2021-12-03
天津医科大学学报(2021年4期)2021-08-21
医学食疗与健康(2021年27期)2021-05-13
健康之家(2020年15期)2020-05-08
中国医学创新(2019年9期)2019-08-19
中国民族民间医药·下半月(2016年8期)2016-10-24
滨州医学院学报(2016年2期)2016-05-27
医学研究杂志(2015年9期)2015-07-01
医学研究杂志(2015年12期)2015-06-10
中国民族民间医药·下半月(2014年5期)2014-12-02
