
基于精准医疗的两例基因突变患儿病例分析
2018-07-02 06:26:20姚献花
中国全科医学 2018年17期
姚献花
在既往的康复工作中,对发育落后、运动障碍、行为异常、认知及语言障碍等患儿的治疗常选用传统的康复治疗方法,如按摩、推拿、针灸、语言认知训练及现代康复训练的运动疗法(PT)、作业疗法(OT)等,部分患儿可取得较好的疗效;但是,对先天因素所导致的发育异常患儿,常效果甚微。随着分子医学技术的发展及广泛应用于临床,这些疑难病症能够给予明确的诊断,并对康复治疗及预后的判断给予正确、精准的指导。本文通过对2例罕见病例的讨论,目的是阐述基因检测在康复医学领域中的重要作用;其意义是建议医生在临床工作中不仅对患者的临床症状、体征及其他实验室检测指标要综合分析,必要时结合基因检测技术以明确诊断、指导治疗及预后评估。
1 病例简介
患儿1,男,1岁2个月,汉族,以发现“下肢不支持体质量3个月”为代主诉于2015-05-06收入河南中医药大学第一附属医院。入院症见:拱背坐,不会爬,不会独站、独走,立位双下肢支持体质量差,纳食可,二便正常。个人史:患儿系G2P2,孕36+5周,顺产,出生体质量2.8 kg,出生后哭声可,出生后病理性黄疸,黄疸于1个月内消退,混合喂养,无易惊、易激惹病史。既往史:出生后有病理性黄疸病史,否认药物、食物过敏史,已按计划进行预防接种。发育史:3个月抬头,8个月坐,8个月翻身。
体格检查:一般体格检查无异常,但患儿四肢及躯干部皮肤干燥粗糙如鱼鳞,手足有大面积脱皮,面部皮肤细白。
专科检查:追视追听可,能笑出声,会无意识发“ba”“o”音。会主动抓物,拇指内收,可拇示指对捏;竖头稳,会主动翻身;坐位拱背坐,坐位侧方平衡(+);俯卧位肘支撑,短时手支持,抬胸,可抬头90°;扶站时双下肢可短时支持体质量,足外翻,下肢稍外旋,屈髋,不会独站,可短时扶栏站立;双上肢肌张力不高,踝膝关节被动活动时稍抵抗,下肢肌张力高,股角110°,腘角130°,足背屈角60°,肌力降低,下肢肌力4+级。原始反射:拥抱反射(-),非对称性紧张性颈反射(ATNR)(-),对称性紧张性颈反射(STNR)(-),侧弯反射(+),紧张性迷路反射(TLR)(-),手握持反射(-),足把握反射(+)。落伞反射(-),颈软无抵抗,双侧膝腱反射亢进,双侧巴氏征(+),踝阵挛(-)。
辅助检查:染色体检查未见异常(2015-03-11外院检查)。
入院诊断:(1)脑性瘫痪;(2)遗传代谢病?(3)鱼鳞病?
诊疗经过:入院后给予综合康复治疗;并于2015-06-29检查颅脑磁共振成像未见异常,2015-12-28遗传代谢病筛查未见典型改变,生物素未见异常;给予生物素口服试验性治疗后患儿皮肤粗糙、双手/足蜕皮现象较前好转,但仍不能站立与行走,下肢肌张力增高,膝腱反射亢进,踝阵挛(+),因治疗时间较长,效果不明显,建议到北京进一步明确诊断,至北京大学第一附属医院检查颅脑磁共振成像未见异常,血常规及生化未见异常,考虑诊为:(1)神经变性病;(2)线粒体病?(3)溶酶体病?建议做基因检查,2016-03-15,由北京福佑龙惠遗传病专科门诊基因检测公司经基因检测后确诊为Sjögren-Larsson综合征。患儿基因检测结果见表1、图1。

表1 患儿1及父母基因检测结果Table 1 Genetic test results of case 1(14-month-old male child) of genic mutation and his parents

图1 患儿1及其父母ALDH3A2基因测序图Figure 1 ALDH3A2 genomic sequence of case 1(14-month-old male child) of genic mutation and his parents
患儿2,男,9个月,因“发现运动发育落后5月余”于2016-05-05入住河南中医药大学第一附属医院。现病史:5个月前(3月龄时)患儿竖头不稳,不会翻身,当时检查颅脑磁共振成像提示蛛网膜下腔增宽,枕大池扩大,查体肋骨外翻,肌力低下,未予重视;4个月前至武汉某医院考虑为戊二酸尿症,检查遗传代谢尿筛查、肌电图未见异常,心肝胆胰脾肾脏彩超未见异常。生化提示肝损伤,给予肌苷片、联苯双酯等保肝降酶药物治疗(1个月后复查降至正常),后至襄阳市某院诊断为中枢性协调障碍,给予综合康复治疗1个月余,患儿仍然竖头不稳,不会翻身,姿势异常,体质量不增;后至北京儿童医院复查磁共振成像结果显示双侧豆状核及尾状核萎缩伴长T2信号,双侧脑室扩张,双侧脑室后部白质T2W信号偏高,脑干听觉诱发电位异常,喉镜提示喉软化症,尿有机酸分析未见异常,血氨基酸提示多种氨基酸降低,丁二酰肉碱增高,提示短链脂肪酸代谢不畅,乳酸、血氨、铜蓝蛋白、巨细胞病毒、EB病毒均阴性,诊断为:(1)发育倒退;(2)肝功异常;(3)先天性喉喘鸣;给予保肝治疗,并行全套基因筛查,家属为求进一步治疗遂至本院。入院症见运动发育落后,竖头不稳,不会翻身,不会坐,双下肢不支持体质量,易惊,喉间痰鸣,四肢姿势不对称,紧张状态下四肢不随意运动更明显,纳眠一般,二便正常。
个人史:患儿系G1P1,孕40周后行剖宫产(因家属考虑妊娠期过长,要求剖宫产),出生体质量3.5 kg,出生过程顺利,无产伤及窒息史,哭声可,新生儿期易惊颤,有哭闹,黄疸持续约1个月。家族史:父母体健;否认家族遗传病病史。父母非近亲结婚。体格检查:意识清,精神可,追视可,追听反应差,可认人,与人交流意识差,表情淡漠,反应迟钝,形体消瘦,面色无华,四肢活动少,未见通贯掌。头围45 cm,前囟1.5 cm×1.5 cm,平软,体质量7.5 kg。眼睑无水肿,无下垂,双侧瞳孔等大等圆,对光反射灵活。气管居中,双肺听诊呼吸音清,未闻及干湿啰音。心率:110次/min,心音有力,律齐,未闻及病理性杂音。腹软,无膨隆,肝脾肋下未触及,肠鸣音正常。肋骨稍外翻,脊柱四肢无畸形,肛门及外生殖器无异常。
专科检查:追视可,追听反应差。双手拇指内收,无主动抓物意识。竖头不稳,不会翻身,不会坐,仰卧位四肢姿势不对称;侧卧位呈角弓反张状,俯卧位肘不支撑,抬头15°~45°;立位下肢不支持体质量;安静状态下四肢肌力、肌张力偏低;双上肢肩肘关节松弛,活动无抵抗,内收角60°~80°,腘角150°~180°,足背屈角30°,四肢肌力约3级。Vojta姿势反射:拉起反射头后坠,无主动抬头意识;立位悬垂反射下肢伸展,双足交叉;俯卧悬垂反射躯干呈倒“U”型。原始反射:拥抱反射伸展相,侧弯反射(+),TLR(+);ANTR(+);STNR(-);手足把握反射(+);双侧膝腱反射可引出,踝阵挛(-),双侧巴氏征(+)。
住院期间完善检查:血常规:白细胞计数5.64×109/L,中性粒细胞分数0.198,淋巴细胞分数0.666;大便常规未见异常,传染病四项未见异常,甲状腺功能未见异常,优生优育十项未见异常。生化指标:血氨41 μmol/L,丙酮酸127 μmol/L,总胆固醇2.73 mmol/L,三酰甘油2.32 mmol/L,丙氨酸氨基转移酶(ALT)49 U/L,天冬氨酸氨基转移酶(AST)65 U/L,肌酸激酶206 U/L,肌酸激酶同工酶32 U/L;铜蓝蛋白0.25 g/L,同型半胱氨酸5.90 μmol/L,乳酸2.10 mmol/L,葡萄糖4.39 mmol/L;25-羟维生素D 39.49 nmol/L。24 h脑电图示:正常婴儿脑电图。基因检测结果显示(北京信诺佰世医学检验所):线粒体DNA耗竭综合征9型(脑肌病型)伴甲基丙二酸尿症。基因测序提示SUCLG1基因发现2个杂合突变点,分别为c.961C>G(p.A321P)和c.713T>C(p.D238 G)(见表2、图2~3),遗传方式为常染色体隐性遗传。其中c.961C>G杂合突变为已报道的致病突变,来自母亲;c.713T>C杂合突变来自父亲,目前未见文献报道,但生物分析软件功能预测显示可能具有致病性。推测可能为我国首例。
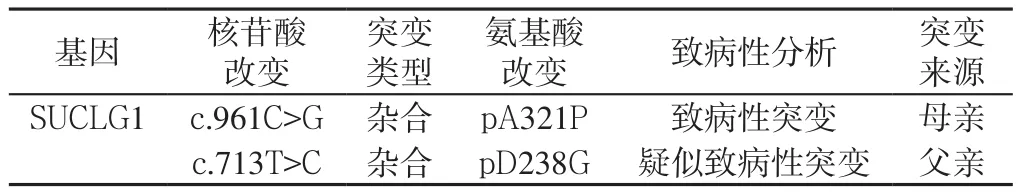
表2 患儿2基因检测结果Table 2 Genetic test results of case 2 (9-month-old male child) of genic mutation
2 讨论
精准医疗是指在分子医学技术的指导下,对某些诊断不明、疗效不好的患儿,通过对DNA的检测帮助医生对疾病做出正确的诊断、治疗及预后判断,从而给患儿明确的治疗方向,避免不必要的治疗,给患儿家庭减轻负担。
本文患儿1根据临床症状及基因检测确诊为Sjögren-Larsson综合征,该病在世界上属于罕见病例,发病率为0.4/100 000[1];是一种罕见的常染色体隐性遗传性神经鱼鳞病样综合征。临床以先天性鱼鳞病、痉挛性双下肢瘫或四肢瘫及智力障碍为特点[2]。对于本病的治疗尚无其他治疗方法,仅有支持疗法对症处理,运动落后者给予康复治疗,长期的理疗对于抵抗痉挛和保存活动能力非常重要[3];但是,康复治疗效果不明显,患儿不能独立行走,只能借助轮椅生活;若无基因检测,患儿常被误诊为痉挛型脑瘫,长期在医院康复治疗,消耗患儿家属大量时间,也造成严重的经济负担;本例患儿明确诊断后,家属放弃在院治疗,转为家庭康复。建议临床若怀疑Sjögren-Larsson综合征时,注意诊断两个要点:皮肤干燥粗糙如鱼鳞和痉挛性瘫痪,早期行基因检测有助于及时发现,明确国内ALDH3A2的突变谱及为早期诊断、遗传咨询产前检查提供依据。目前基因治疗还存在许多问题,尚在试验探索阶段,然而基因治疗是将来实现根治的期望。

图2 患儿2及其父母SUCLG1基因测序图Figure 2 SUCLG1 genomic sequence of case 2 (9-month-old male child) of genic mutation and his parents shows the compound heterozygous for a mutation c.961C>G(p.A321P)coming from the mother

图3 患儿2及其父母SUCLG1基因测序图Figure 3 SUCLG1 genomic sequence of case 2 (9-month-old male child) of genic mutation and his parents shows the compound heterozygous for a mutation c.713T>C(p.D238 G)coming from the father
本文患儿2确诊为线粒体DNA耗竭综合征9型,其发病率至少为1/5 000[4],属于线粒体疾病的一个类型;儿童线粒体脑病临床表现多样,容易误诊,陈健等[5]报道该院在2008—2013年共收治11例线粒体脑病患儿,其中7例曾被误诊,误诊率极高;其主要原因是不同患儿的同一点突变可出现不同的异常临床症状[6];本例患儿早期曾误诊为脑损伤、脑性瘫痪,分别在多家医院康复治疗,疗效不好,最后经基因检测确诊为线粒体DNA耗竭综合征9型。
线粒体DNA耗竭综合征临床上分为肌病、脑肌病、肝性脑病和神经胃肠道疾病4种,目前发现由9种维持线粒体DNA稳定性的核基因突变所致[7];其中脑肌病型(SUCLA2、SUCLG1、RRM2B)SUCLA2缺陷,全球约报道20例患儿。关于SUCLG1缺陷的研究较少,临床表现与SUCLA2缺陷类似,但症状较重[8]。线粒体DNA耗竭综合征9型由SUCLG1基因突变引起,以常染色体隐性方式遗传,该型仅伴有甲基丙二酸尿症,是由于SUCL缺陷,不能催化琥珀酰CoA生成琥珀酸,造成琥珀酰CoA堆积,同时使甲基丙二酰CoA生成琥珀酰CoA的过程受阻,导致甲基丙二酸升高[7];该患儿携带SUCLG1基因的两个杂合突变:c.961C>G(p.A321P)和c.713T>C(p.D238G),其中c.961c>G(p.A321P)为文献报道的已知致病性突变[8],而c.713T>C(p.D238G)未见文献报道,因此,SUCLG1基因c.961C>G(母)和c.713T>C(父)2个杂合突变可能为我国首例,该报道扩展了SUCLG1基因突变谱,为患儿病因诊断及该家系的遗传咨询和产前诊断提供了分子依据。
总之,在既往的临床工作中遗传代谢性疾病多见,多以运动落后,发育迟缓,运动姿势异常,语言障碍,癫痫,反复呕吐或腹泻,体质量不增,肌张力障碍等症状而就诊,若无基因检测技术协助诊断,患儿难以明确诊断,仅是常规的康复手段治疗,对一般发育落后,脑性瘫痪患儿来说,可以取得较好疗效;而对于遗传代谢性疾病来说,诊断难以明确,治疗效果甚微,甚至延误病情;目前随着医学的发展,分子遗传学已被广泛应用于临床,某些患有遗传代谢性疾病的患儿能够得到正确的诊断与治疗,同时也给患儿明确的预后判断;因此,基因检测技术在临床中显得尤为重要,能够为更多的患儿获得准确的治疗和优质的服务。
本文无利益冲突。
[1]高蕾,蒋丽琼,石秀玉,等.小儿Sjögren-Larsson综合征临床特征及基因突变分析[J].中国实用儿科杂志,2014,29(6):458-461.DOI:10.7504/ek2014060614.GAO L,JIANG L Q,SHI X Y,et al.Analysis of clinical manifestations and gene mutations of Sjögren-Larsson syndrome[J].Chinese Journal of Practical Pediatrics,2014,29(6):458-461.DOI:10.7504/ek2014060614.
[2]许汝钗,宋兰萍,张英,等.Sjogren-Larsson综合征一家系二例报告[J].中国优生与遗传杂志,2004,12(5):129.DOI:10.3969/j.issn.1006-9534.2004.05.075.XU R C,SONG L P,ZHANG Y,et al.Two cases report of Sjogren-Larsson syndrome[J].Chinese Journal of Birth Health and Heredity,2004,12(5):129.DOI:10.3969/j.issn.1006-9534.2004.05.075.
[3]张轶凡,黄坚.鱼鳞样红皮症-痉挛性截瘫-智力发育不全综合征(Sjogren-Larsson综合征)(附1例报道及文献分析)[J].齐齐哈尔医学院学报,2013,34(22):3335-3336.ZHANG Y F,HUANG J.Sjogren-Larsson syndrome(one case report and literature review)[J].Journal of Qiqihar University of Medicine,2013,34(22):3335-3336.
[4]刘志梅,方方,丁昌红,等.二代测序在儿童线粒体病诊断中的应用[J].中华儿科杂志,2015,53(10):747-753.DOI:10.3760/cma.j.issn.0578-1310.2015.10.009.LIU Z M,FANG F,DING C H,et al.Diagnosis of mitochondrial disorders in children with next generation sequencing[J].Chinese Journal of Pediatrics,2015,53(10):747-753.DOI:10.3760/cma.j.issn.0578-1310.2015.10.009.
[5]陈健,邹丽萍.儿童线粒体脑病的临床和分子遗传学特点及其预后[J].临床儿科杂志,2014,32(11):1020-1023.DOI:10.3969/j.issn.1000-3606.2014.11.006.CHEN J,ZOU L P.Clinical and molecular-genetic features and prognosis of mitochondrial encephalopathy in children[J].Journal of Clinical Pediatrics,2014,32(11):1020-1023.DOI:10.3969/j.issn.1000-3606.2014.11.006.
[6]黄圆圆.线粒体疾病研究进展[J].国际儿科学杂志,2013,40(5):505-510.DOI:10.3760/cma.j.issn.1673-4408.2013.05.019.HUANG Y Y.Research progress of mitochondrial disease[J].International Journal of Pediatrics,2013,40(5):505-510.DOI:10.3760/cma.j.issn.1673-4408.2013.05.019.
[7]刘志梅,方方,丁昌红,等.SUCLA2相关脑肌病型线粒体DNA耗竭综合征一例并文献复习[J].中华儿科杂志,2014,52(11):817-821.DOI:10.3760/cma.j.issn.0578-1310.2014.11.005.LIU Z M,FANG F,DING C H,et al.SUCLA2-related encephalomyopathic mitochondrial DNA depletion syndrome:a case report and review of literature[J].Chinese Journal of Pediatrics,2014,52(11):817-821.DOI:10.3760/cma.j.issn.0578-1310.2014.11.005.
[8]刘玉鹏,李溪远,丁圆,等.琥珀酰辅酶A连接酶缺陷导致继发性甲基丙二酸尿症四例的临床与实验室研究[J].中华 儿 科 杂 志,2016,54(5):365-368.DOI:10.3760/cma.j.issn.0578-1310.2016.05.011.LIU Y P,LI X Y,DING Y,et al.Clinical and laboratory studies on four Chinese patients with succinate-CoA ligase deficiency noticed by mild methylmalonic aciduria[J].Chinese Journal of Pediatrics,2016,54(5):365-368.DOI:10.3760/cma.j.issn.0578-1310.2016.05.011.
猜你喜欢
区域治理(2022年40期)2022-11-27 04:01:54
中国临床医学影像杂志(2022年5期)2022-07-26 07:12:00
海洋通报(2021年1期)2021-07-23 01:55:14
中医眼耳鼻喉杂志(2021年2期)2021-07-21 08:53:40
生物学通报(2021年4期)2021-03-16 05:41:26
儿童故事画报(2020年12期)2020-06-23 06:48:16
动漫界·幼教365(小班)(2019年10期)2019-10-28 02:04:20
动漫界·幼教365(大班)(2019年10期)2019-10-28 01:54:09
动漫界·幼教365(中班)(2019年10期)2019-10-28 01:53:17
猪业科学(2018年8期)2018-09-28 01:27:32
