
原发性皮肤边缘区B细胞淋巴瘤一例并文献复习
2018-07-02 01:26孔泽琳颜潇潇陈声利卢宪梅刘永霞周桂芝王广进田洪青
中国麻风皮肤病杂志 2018年6期
孔泽琳 颜潇潇 陈声利 卢宪梅 刘永霞 周桂芝 郭 璇 王广进 田洪青
原发性皮肤边缘区B细胞淋巴瘤(primary cutaneous marginal zone B-cell lymphoma, PCMZL)是原发性皮肤B细胞淋巴瘤中最常见的一种,恶性程度低,属惰性淋巴瘤。WHO分类中其被列为结外边缘区黏膜相关组织淋巴瘤(MALT),1983年Isaacson和Wright首先将MALT作为一种特殊类型的淋巴瘤进行叙述[1]。但两者之间的关系存在争议,有学者认为应将其看做一个独立病种[2]。PCMZL包括以往的原发性皮肤免疫细胞瘤、伴单克隆浆细胞的皮肤滤泡淋巴样增生及部分皮肤髓外浆细胞瘤[3]。临床表现为单发或多灶性斑块或结节,组织病理由小的边缘区B细胞、淋巴细胞、浆细胞及反应性T细胞混合浸润组成,特征性表达Bcl-2,缺乏Bcl-6和CD10表达[4]。本文将报道1例PCMZL并将国内外近年来关于此病的研究报道进行回顾复习,以加深对该病的认识,为临床诊疗提供一定的依据。
1 临床资料
患者,女,59岁。因右股部斑块3年就诊。患者3年前无明显诱因于右股部出现红色斑块,自觉轻度肿胀,无疼痛、瘙痒等自觉症状,曾于当地按“皮肤感染”治疗,效果欠佳,遂至我院门诊就诊。患者自发病以来,饮食睡眠精神可,体重未见明显减轻。既往体健,否认系统性疾病史、传染病史、药物食物过敏史及家族中其他成员的类似病史。体格检查:全身浅表淋巴结未触及肿大,各系统检查未见异常。皮肤科检查:于右股部见一类椭圆形淤紫色斑块,皮损触之质硬,有浸润感,边界清楚(图1)。临床拟诊:蕈样肉芽肿?皮肤转移癌?辅助检查:血生化:乳酸脱氢酶278 U/L,肌酸激酶同功酶26 U/L,α-羟丁酸脱氢酶231 U/L。心电图未见明显异常。胸部、全腹部CT均未见明显异常。组织病理示:(右股部)表皮大致正常,表皮下见一无浸润带,真皮全层片状淋巴样细胞浸润(图2)。免疫组化:淋巴样细胞CD20(+)、CD79a(+)、Bcl-2(+)、MUM-1部分细胞(+)、小淋巴细胞CD3(+)、CD4(+)、CD5(+)、CD21示不规则FDC网、少许浆细胞CD38(+)、Kappa(-)、Lambda部分(+),呈单克隆性表达,CD10、Bcl-6、CD30、CD56、CD123、cyclingD1均阴性,Ki-67约30%~40%(+)(图3)。原位杂交:EBER阴性。诊断:符合皮肤边缘区B细胞淋巴瘤。建议患者于综合医院血液科或肿瘤科就诊系统治疗,患者外科切除治疗,随访1年多病情未复发,且无新发皮损。
2 讨论
1982年公布的工作规范(working formulation,WF)及1988年修订的Kiel分类是较早的关于淋巴瘤的分类方案,但并不适用于临床,1994年发表修正的欧美淋巴瘤分类是较早的科学全面的恶性淋巴瘤分类。上述三种分类中均包括边缘区淋巴瘤[5]。2005年世界卫生组织-欧洲癌症研究与治疗组织(WHO-EORTC)对原发性皮肤B细胞淋巴瘤的定义为:发病仅有皮肤表现,并且病情进展6个月内未出现皮肤以外其他脏器转移的B细胞淋巴瘤[6]。根据2008年WHO-EORTC分类标准,又分为3种主要类型:①原发性皮肤滤泡中心淋巴瘤(PCFCL);②原发性皮肤边缘区淋巴瘤(PCMZL);③原发性皮肤弥漫大B细胞淋巴瘤/腿型(PCDLBCL/LT)。国内明确诊断为原发性皮肤边缘区B细胞淋巴瘤的病例报道较少[7-10],大约有4例,不包括未明确分型的原发性皮肤B细胞淋巴瘤,其中男2例,女2例,平均发病年龄为(43.00±12.36)岁,其中病程最长的1例为30余年,最短的1例为1年。国外对该病的报道及研究较多并且相对成熟。

图1 右股部淤紫色斑块
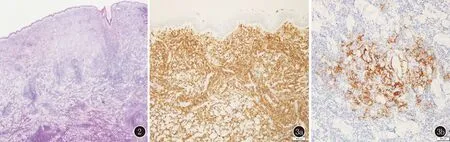
图2 表皮大致正常,表皮下见一无浸润带,真皮全层片状淋巴样细胞浸润(HE,×40)图3 3a:CD20肿瘤细胞阳性(SP,×100);3b:CD21不规则FDC网(SP,×200)
边缘区淋巴瘤与3号和(或)18号染色体三倍体及t(11;18)(q21;q21)易位有关。t(11;18)(q21;q21)易位是其特征性遗传学标志,此易位可引起11号染色体上凋亡抑制因子-2(API2)与18号染色体上的MALT1基因相融合[11]。也有许多患者存在(14;18)(q32;q21)易位,该染色体易位可使Bcl-2免疫球蛋白重链基因(IgH)增强子调控导致重排,使其在肿瘤细胞中持续表达,从而抗凋亡[12]。t(1;14)(p22;q32)导致Bcl-10表达异常与MALT淋巴瘤发病亦有关,上述易位可导致NF-κB分子通路激活,亦是MALT淋巴瘤的发病机制之一[13]。与其他MALT淋巴瘤不同,PCMZL的发病与自身免疫性疾病无关,部分患者发病可能与感染伯氏疏螺旋体有关[3],但可能只限于欧洲部分区域。
PCMZL皮损表现为无症状的丘疹、结节或斑块,红、棕红或紫色,周围可绕以浸润性红斑,皮损常单发,好发于躯干、四肢[14],平均发病年龄约50岁。关于PCMZL与其他疾病的关联,有国外一项对80例PCMZL患者进行的病例对照研究,发现有高达61.2%的PCMZL患者存在胃肠道疾病,较对照组的37.5%有明显差异,一半的PCMZL患者存在胃食管反流,20%的PCMZL患者幽门螺杆菌血清学检查阳性。包括肠易激综合征及炎症性肠病在内的结肠疾病较对照组更多见。另外20%的PCMZL患者存在不同的自身免疫性疾病,包括桥本甲状腺炎、红斑狼疮等[15]。
MALT淋巴瘤的组织病理学共同特点包括反应性淋巴滤泡、淋巴上皮病变和几种特异性细胞成分[11]。PCMZL病理表现为真皮及皮下组织的浸润,浸润的肿瘤细胞通常由小淋巴细胞、边缘区B细胞、浆细胞样淋巴细胞、成熟的浆细胞和反应性生发中心及巨噬细胞构成[2],生发中心通常呈浅灰色,有多形性增生,有时还可见少量中心母或免疫母样细胞及反应性T细胞[11,16],某些情况下反应性T细胞甚至可完全遮盖肿瘤B细胞。淋巴瘤细胞可以含有“Dutcher小体”(嗜酸性核内包涵体),即免疫球蛋白核内聚集。PCMZL往往在表皮下方向真皮内呈楔形生长,具有“头重”的特点,肿瘤常形成结节状,这些结节部分是反应性淋巴滤泡,部分是滤泡植入后形成的肿瘤性结节[11]。PCMZL被认为具有两个亚型,其中常见的一种为较常见的转化型,表达IgG、IgA、IgE的浆细胞和许多混合T细胞浸润在血管及肿瘤结节周围,且存在表达IgD的反应性生发中心,肿瘤细胞缺乏CXCR3的表达。另一种亚型为少见的非转化型,其特征在于浸润细胞一半为的表达IgM和CXCR3的肿瘤B细胞而反应性T细胞较少,此类肿瘤B 细胞其亦是干扰素γ诱导的趋化因子受体,这也是其他MALT淋巴瘤的共同特征[16]。免疫表型为肿瘤细胞CD20+、CD79a+、bcl-2+、mum-1(+),CD5-、CD10-、CD23-,生发中心标志物Bcl-6和CD10在肿瘤细胞中呈阴性,但如果存在反应性淋巴滤泡则呈阳性[3]。浆细胞限制性表达轻链蛋白kappa,有研究发现PCMZL存在较高的IgG4表达率[17]。仅有部分PCMZL病例可见增多的大细胞,且极少转化为大细胞淋巴瘤。CD5、CyclinD1等免疫组化染色可与淋巴瘤继发于皮肤相鉴别[4]。有国外研究认为,浆细胞样树突状细胞(PDC)在启动T细胞及B细胞的免疫应答和激活中起到关键作用,CD123+的FDC在PCMZL的早期病变和初始复发中有大量存在,而在其他B细胞淋巴瘤中存在较少,因此在PCMZL中检测PDC簇可作为有用的辅助诊断标记[18]。
国际皮肤淋巴瘤协会(ISCL)及欧洲癌症研究与治疗组织(EORTC)建议对CBCL进行临床分期,主要依据包括病史、查体、实验室检查及全身CT或PET检查。PCMZL无需进行骨髓穿刺及活检[3]。对于PCMZL,可应用聚合酶链式反应(PCR)检测免疫球蛋白(Ig)轻链的单克隆表达或Ig重链的克隆重排,但国外有病例报道过双克隆多病灶的PCMZL[19]。
PCMZL的鉴别诊断包括:①弥漫型皮肤滤泡中心淋巴瘤:肿瘤细胞浸润主要为中心细胞及中心母细胞,因CD10和Bcl-6是生发中心细胞的标志物,故该病表达CD10及Bcl-611;②B细胞慢性淋巴细胞白血病(B-CLL)也累及皮肤,临床上及组织学上都与PCMZL相似,但肿瘤细胞所表达的免疫表型不同,肿瘤细胞CD20+,CD79a+,CD5+,CD23+和CD43+2;③皮肤淋巴组织增生(CLP):浸润细胞成分复杂,除小淋巴细胞,还可见组织细胞等其他炎细胞[20],且皮损常见于头部,尤其是耳垂。
考虑到PCMZL为惰性淋巴瘤,如患者皮损表现为独立的结节或累及部位较少,可进行局部放疗或切除[4]。如有伯氏疏螺旋体感染血清学阳性或应用PCR技术检测到伯氏疏螺旋体阳性的病例,可以尝试给予口服抗生素(头孢菌素类或四环素类)。皮损内注射干扰素-α、利妥昔单抗、糖皮质激素均可作为二线治疗方案[2]。多发皮损可给予苯丁酸氮芥、干扰素α或利妥昔单抗治疗,国内有报道一例皮损泛发并伴有多发淋巴结的PCMZL应用R-CHOP方案化疗,其中利妥昔单抗用量为600 mg,效果显著[10]。国外曾报道一例颅内受累的PCMZL,对利妥昔单抗反应良好,治疗8年后无皮外转移[21]。电化学疗法可作为一种治疗PCMZL的新方法,即将电穿孔和化疗药物组合用于局部治疗,电穿孔可使电脉冲通过孔道诱导细胞膜的瞬时透化,从而使化疗药物扩散到胞质溶胶中,电化学疗法对全身脏器毒副作用低,可用于姑息性治疗不能切除的复发性PCMZL结节,不良反应为色素沉着[22]。
该病预后良好,5年生存率约为100%[11],预后明显好于其他部位的MALT淋巴瘤[23],但该病复发率较高,可达50%,少有远处脏器转移[3],仅有<10%的患者可见皮下扩散,尤其是非转移型亚型[2],少有骨髓受累。远处播散通常发生于大细胞转化及t(14;18)(q32;q21)Ig H/Bcl-2和t(14;18)(q32;q21)Ig H/MALT1的易位[2]。国际结外淋巴瘤工作组(IELSG)提出与惰性皮肤淋巴瘤预后相关的独立危险因素,包括①乳酸脱氢酶(LDH)升高;②累及皮肤部位大于2处;③结外其他病变;据研究统计,以上危险因素均不具备时,疾病5年无进展生存率可高达91%,如存在2个或以上危险因素时,5年无进展生存率只有48%[24]。另外,MYD88L265P基因突变也与预后相关[4]。
本例患者具有以下特点:①中老年女性,病史3年,皮损表现为单发的右股部淤紫色斑块。②组织病理表现典型,免疫组化支持皮肤边缘区B细胞淋巴瘤。③相关辅助检查排除系统淋巴瘤累及皮肤的可能性,结合临床表现、病史及组织病理等可明确诊断为原发性皮肤边缘区B细胞淋巴瘤。④该患者具备一项危险因素,即LDH升高,预后应较良好。
[1] 邱丙森.原发性皮肤B细胞淋巴瘤的研究进展(二)[J].临床皮肤科杂志,2000,29(5),315-317.
[2] Selva RL, Violetti SA, Delfino C, et al. A literature revision in primary cutaneous B-cell lymphoma[J]. Indian J Dermatol,2017,62(2):146-158.
[3] Hope CB, Pincus LB. Primary cutaneous B-cell lymphomas[J]. Clin Lab Med, 2017, 37:547-574.
[4] Wilcox RA. Cutaneous B-cell lymphomas:2016 update on diagnosis,risk-stratification,and management[J].Am J Hematol,2016,91(10):1052-1055.
[5] 邱丙森.皮肤恶性淋巴瘤的分类:近代提案的分析和评价[J]. 临床皮肤科杂志,2000,29(4):242-244.
[6] Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas[J]. Blood,2005,105:3768-3785.
[7] 周文明,汪宇,张伟,等.原发性皮肤边缘区B细胞淋巴瘤1例并文献复习[J]. 贵州医药,2017,41(9):973-975.
[8] 陈字,陈彤,谢彦晖,等.原发性皮肤边缘区B细胞淋巴瘤1例[J]. 临床血液学杂志,2009,22(1):50-51.
[9] 钟伟龙,窦侠,叶庭路,等.原发性皮肤边缘区B细胞淋巴瘤[J]. 临床皮肤科杂志,2017,46(5):341-344.
[10] 毛笑非,闫言,孙秋宁.原发性皮肤边缘区B细胞淋巴瘤1例[J]. 协和医学杂志,2012,3(4):487-489.
[11] 李白周,孔藴毅,陆洪芬,等.原发性皮肤边缘区B细胞淋巴瘤组织病理特征和免疫表型分析[J].临床皮肤科杂志,2007,36(12):750-753.
[12] James O, Randy D, Gascoyne MD, et al. The lancet:Non-Hodgkin lymphoma[J]. Lancet,2017,7:298-310.
[13] 王筱璨,克晓燕. MALT淋巴瘤病因及发病机制研究进展[J].中国实验血液学杂志,2012,20(6):1526-1530.
[14] Kempf W, Kazakov DV, Buechner SA, et al. Primary cutaneous marginal zone lymphoma in children: a report of 3 cases and review of the literature[J]. J Am Dermatopathol,2014,36 (8):661-666.
[15] Guitart J, Deonizio J, Bloom T, et al. High incidence of gastrointestinal tract disorders and autoimmunity in primary cutaneous marginal zone B-Cell lymphomas[J]. JAMA Dermatol,2014,5:E1-E7.
[16] Edinger JT, Kant JA, Swerdlow SH. Cutaneous marginal zone lymphomas have distinctive features and include 2 subsets[J]. Am J Surg Pathol,2010,34:1830-1841.
[17] Brenner I, Roth S, Puppe B, et al. Primary cutaneous marginal zone lymphomas with plasmacytic differentiation show frequent IgG4 expression[J]. Modern Pathology,2013,26:1568-1576.
[18] Kutzner H, Kerl H, Monique C, et al. CD123-positive plasmacytoid dendritic cells in primary cutaneous marginal zone B-cell lymphoma diagnostic and pathogenetic implications[J]. Am J Surg Pathol,2009,33(9):1307-1313.
[19] Nicholson KM, Patel KP, Duvic M, et al. Bi-clonal, multifocal primary cutaneous marginal zone B-cell lymphoma: report of a case and review of the literature[J]. J Cutan Pathol,2012,39:866-871.
[20] Tsuji K, Suzuki D,Naito Y, et al. Primary cutaneousmarginal zone B-cell lymphoma[J]. Eur J Dermatol,2005,15(6):480-483.
[21] Virmani P, Busam K, Patricia L, et al. Primary cutaneous marginal zone lymphoma with leptomeningeal involvement and a durable response to rituximab[J]. JAAD Case Reports,2017,3:269-272.
[22] Gatti A, Stinco G, Trevisini S, et al. Electrochemotherapy as a novel treatment for primary cutaneous marginal zone B-cell lymphomas[J]. Dermatol Ther,2014,27:244-247.
[23] Olszewski AJ, Castillo JJ. Survival of patients with marginal zone lymphoma: analysis of the surveillance, epidemiology, and end results database[J]. Cancer,2013,119(3):629-638.
[24] Ryan A. Wilcox. CME Information: Cutaneous B-cell lymphomas: 2016 update on diagnosis, risk-stratification, and management[J]. Am J Hematol,2016,91(10):1052-1055.
猜你喜欢
中国卫生标准管理(2022年21期)2023-01-03
传染病信息(2022年3期)2022-07-15
中国临床医学(2022年3期)2022-07-08
临床超声医学杂志(2022年4期)2022-05-07
天津医科大学学报(2021年3期)2021-07-21
医学信息(2021年5期)2021-03-21
中国临床医学影像杂志(2019年6期)2019-08-27
中国临床医学影像杂志(2019年5期)2019-08-27
中华老年多器官疾病杂志(2016年9期)2016-04-28
医学研究杂志(2015年2期)2015-06-10
