
高温金属熔体黏度突变探索∗
2018-06-14 08:49:34商继祥赵云波胡丽娜
物理学报 2018年10期
商继祥 赵云波 胡丽娜
(山东大学,材料液固结构演变与加工教育部重点实验室,济南 250061)
1 引 言
高温金属熔体黏度作为描述液体动力学性质的物理参量,体现了液体结构的演变过程,始终受到广泛的关注.在传统铸造工艺中,高温金属熔体黏度变化是决定晶态铸件成型能力和气泡或非金属夹杂物能够顺利排出的关键因素[1].对于利用超快速冷却技术制备的金属玻璃而言,高温金属熔体黏度大小更是决定该合金能否形成金属玻璃或者玻璃形态的要素之一.
1985年,美国科学家Angell[2]首先用液体的脆性概念表征了黏度随温度变化的难易程度.当过冷液体的黏度随温度的变化较大时,即液体的结构随温度的变化容易发生变化时,液体是脆性的,在Angell图上表现为一条曲线;当过冷液体的黏度随温度的变化很难发生改变时,即液体结构不容易发生变化,为强性液体,在Angell图上表现为一条直线.借用该过冷液体的脆性定义,Bian等[3]采用液相线约化温度来表示高温金属熔体黏度在液相线附近变化的难易程度,提出了过热熔体的脆性概念,并发现该脆性大小与熔体中团簇的稳定性密切相关.黏度在衡量金属玻璃形成能力方面也有一定的参考性,玻璃形成能力强的合金体系往往有着较大的黏度值.这是由于大的黏度值可以抑制原子或分子定向移动从而抑制晶化过程,降低了结晶的可能性,从而有利于金属玻璃的形成.例如,在共晶凝固的过程中,当温度降低到共晶点温度时,液相中会同时析出两种不同成分的固溶体相,直至凝固结束.两种不同成分固溶体的形核,以及形核后的晶体长大过程都与金属原子的移动速率或扩散行为密切相关.当金属原子的迁移因较大的黏滞力受阻后,熔体能在较低的温度下仍保持原子排列的无序状态,从而有利于非晶的形成.以往的研究工作已经表明过热熔体的脆性系数与金属玻璃形成能力的大小存在着负相关性[3,4],黏度的变化特征对合金熔体玻璃形成能力的影响举足轻重.金属玻璃形成能力的大小对拓宽金属玻璃的应用范围,实现金属玻璃从功能材料向结构材料质的跨越起着关键性的作用.通常认为,高温金属熔体的黏度在熔点以上随着温度的变化规律符合Arrhenius公式.但越来越多的实验结果表明,高温金属熔体的动力学性质存在转折.高温金属熔体的黏度在很多情况下不能用同一条Arrhenius曲线进行很好的拟合:在特定的温度处黏度偏离Arrhenius曲线,并在偏离的基础上继续符合另外一条新的Arrhenius曲线.这种黏度突变的行为是液-液相变的直观体现,液-液相变现象的发生势必伴随着熔体中金属原子的重组,即熔体中原子或原子团簇从一种状态转变为另一种状态.与固态相变类似,液-液相变也伴随着体积、熵或焓的变化.加深液-液相变深层次的研究,对深入认识液态金属的性质着重要的理论意义.而对于该现象的揭示,其动力学性质即黏度随温度的变化可以给我们提供明显的论证依据.同时,高温金属熔体黏度的突变也极可能是金属玻璃固体异常弛豫行为的根源.
因此,对高温金属熔体黏度突变的探索研究,不仅有助于认识高温金属熔体的动力学性质的演变规律,而且对它的探索有助于进一步认识合金体系的金属玻璃形成能力和液-液相变规律,并将高温合金熔体的动力学性质与合金体系的势能图谱、固体的不同弛豫行为相联系[3−6].这不仅对实际工业生产具有指导意义,同时对理解熔体过-冷液体-金属固体的遗传特征也具有重要的理论价值.这也是今后金属熔体动力学性质研究的重要发展方向之一.
2 高温金属熔体的黏度测量原理
黏度的物理意义定义为:在单位速度梯度下,作用在单位接触面积上的内摩擦力.

式中η表示为动力学黏度,F为内摩擦力,A为内摩擦力与两液体层接触面的面积,dv/dy为速度梯度.如图1所示,设距离为dy的相邻液体层以不同的速度v和v+dv的速度流动,即垂直于剪切力方向产生速度梯度,则在这两液层之间,将产生一对大小相等而方向相反的内摩擦力,力图阻止两液层发生互相运动,产生的这一阻力就是液体的黏滞力.
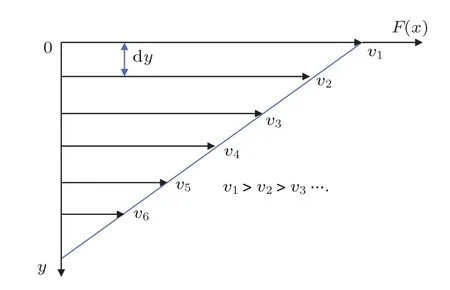
图1 黏度定义示意图Fig.1.Diagram of viscosity definition.
虽然用于测量黏度的方法有很多(如毛细血管法、落体法、旋转法、声波法等),但适合高温金属熔体黏度测量的方法并不多.这往往是由于高温金属熔体的黏度比较小,即使含有稀土元素等大分子量的Fe基、Co基等体系,黏度也不过只有十几mPa·s左右,而且高温金属熔体黏度的测量是在高温下进行的,极易被氧化,因此一些常规的测量黏度的方法并不适合高温金属熔体黏度的测量.目前用于测量高温金属熔体黏度的方法主要是震荡法.基于震荡法的黏度仪有两种形式,一种是悬挂式[7−12],一种是站立式[13].相比于站立式,悬挂式更容易操作和制作,所以实验室中测量高温金属熔体的黏度仪主要是悬挂式.悬挂式黏度仪的结构如图2(a)所示,金属熔体置于圆柱形坩埚内,坩埚由悬丝悬挂,悬丝与步进电机相连,单片机控制步进电机的驱动器,使步进电机转动,从而使悬丝带动坩埚沿垂直轴旋转,在悬丝恢复力的作用下,坩埚做震荡运动,由于液体黏滞的作用,震荡运动逐渐衰减,但运动周期保持不变,在悬丝放置一个反光镜,用于反射激光,计算运动周期和对数衰减率.测量系统如图2(b)所示,A,B两点为光敏光电池用于接受反射激光,在开始测量之前需要将反射光电置于A点,步进电机转动之后,反射激光在A和B两光电池之间来回摆动,光电池通关光电转换效应,计算出激光扫过AA,AB,BA,BB的时间,这样便可以算出周期S和对数衰减率σ,再通过Shvidkovskiy公式[14]计算出黏度值.
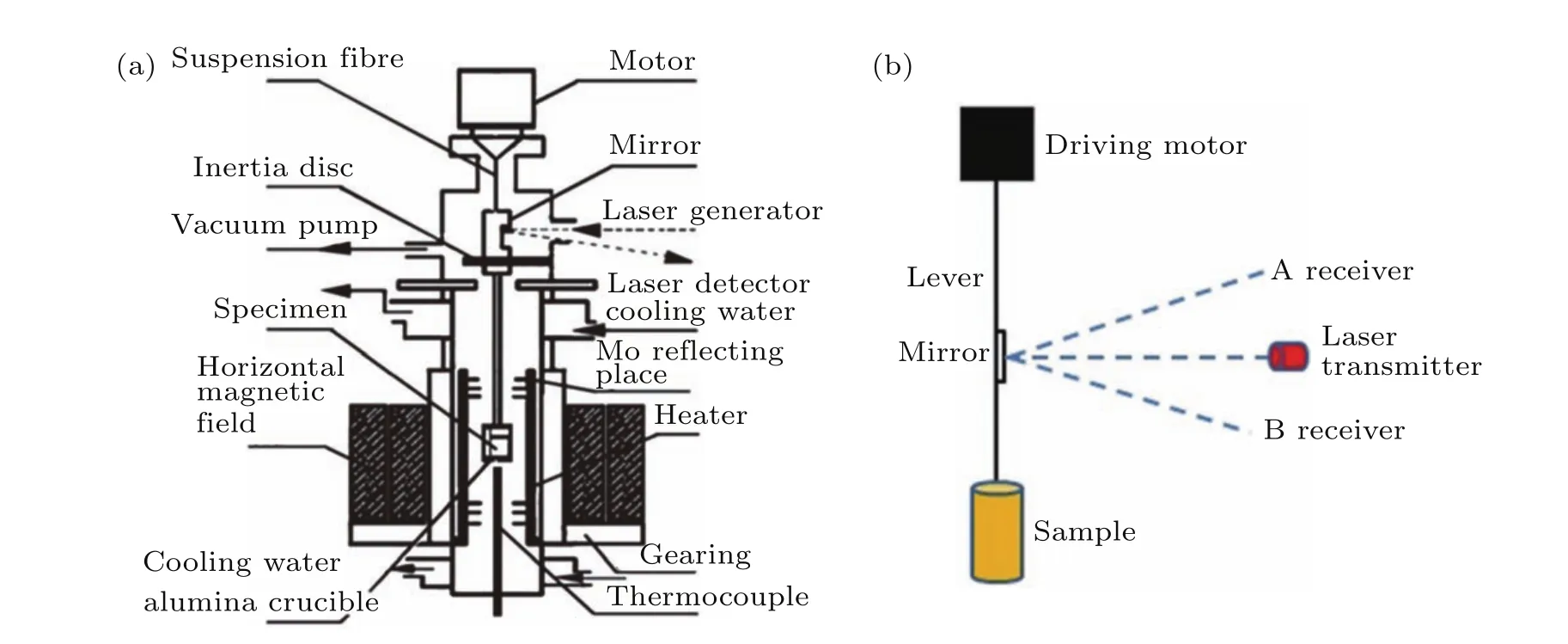
图2 (a)黏度仪示意图[15];(b)测量系统示意图[15]Fig.2.(a)Schematic of viscometer[15];(b)schematic of measuring system[15].
震荡运动的方程为:
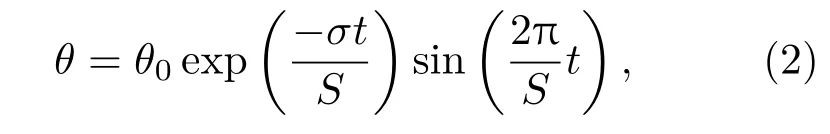
式中θ0是前指因子,S为震荡周期,σ为对数衰减率,t为时间.
Shvidkovskiy公式计算黏度:

式中I表示转动惯量,m表示测试样品的质量,R表示为坩埚的内径,S0表示空坩埚震荡运动周期,σ0表示空坩埚的对衰减率,W是矫正系数,υ表示为运动学黏度,ρ为测试样品的密度.
不同的实验条件和试验方法对黏度测量结果影响较大.以前工作已经表明磁场强度、坩埚表面粗糙度、保温时间以及放料的多少对黏度有着重要的影响.Xu等[15]报道了Ga80Cr20和Ga80Ni20在不同磁场强度下黏度的变化规律,实验结果表明:随着磁场强度不断增加,熔体的黏度不断增加.这是由于高温金属熔体的团簇带有一定量的电荷,带电离子在磁场中会受到洛伦兹力,洛伦兹力改变团簇的运动路径,从而使团簇的运动更为复杂,高温金属熔体的黏滞力增加.磁场强度H越大,黏度值也越大.Wu等[16]报道了测量黏度时所用坩埚的表面粗糙程度对Pb38.1Sn61.9高温金属熔体黏度的影响,实验结果表明:坩埚的表面粗糙程度对黏度有一定的影响,且表面粗糙度越大,在同一温度下测得的黏度值越大.这是因为摩擦力的增加导致对数衰减率δ的增加,黏度也相应地增大.Sun等[17]的工作也证实了坩埚表面的粗糙程度对水的黏度具有正相关的关系:表面粗糙程度越大,水的黏度也越大.放料的多少对黏度影响也比较大[18]:当样品体积较小时,测量的黏度数据大于标准数值,随着体积的增大,黏度数值减小,逐渐接近标准值;当体积继续增大时,黏度数值进一步降低,低于标准值.这是因为样品过多,在振荡运动过程中会溢出,造成黏度测量结果的误差.在进行高温金属熔体黏度测量时,金属熔体体积会发生膨胀,所以体积也因此不宜过大.保温时间的长短同样对黏度有较大的影响,保温初期,合金熔体黏度随保温时间变化会有所波动,但随着保温时间延长,合金熔体黏度波动逐渐减小.为了得到尽可能准确的黏度值,保温时间应控制在30 min以上[19].造成黏度随保温时间变化的原因可以从两个角度来考虑:首先在保温初期控制温度和炉内的温度是不同步的,随着保温时间的增加,由于热辐射的影响,内层熔体的温度逐渐与坩埚套的温度趋于一致,黏度值趋于稳定.其次在保温初期,熔体经过前期温度的变化,原子运动相对比较剧烈,合金熔体的成分相对而言不均匀,合金熔体需要一定的时间达到均匀态,这就导致测量结果随保温时间略有波动.
3 高温金属熔体的黏度
3.1 纯组元单质的黏度
目前,对金属单质高温金属熔体的黏度研究主要集中在较低熔点、不易氧化的元素种类上,对稀土基易氧化金属以及Zr等高熔点金属的黏度研究开展得很少.这主要是受到实验条件(如真空度、最大温度测量范围)的限制.一般认为,高温金属熔体的黏度在熔点以上随着温度的降低,黏度值不断增大,并符合Arrhenius公式,其中η0是前指因子,E是流体团簇的激活能,k为玻尔兹曼常数,T为温度.但相当一部分实验结果表明在整个温度范围内,高温金属熔体的黏度随温度的变化并不是单调的,在某些特定的温度附近,黏度存在突变现象,并在新变化的基础上继续符合Arrhenius公式.越来越多的研究结果显示:高温金属熔体的黏度突变是一普遍的现象,而这一现象与熔体中团簇的变化密切相关.孙民华等[22]报道了Al熔体的黏度,研究了Al熔体黏度随温度的变化规律,发现在升温过程中熔体黏度值在780◦C和950◦C左右发生突变;在降温过程中,黏度的突变发生在930◦C与750◦C,如图3所示.孙民华等通过Al熔体氢含量的测定以及分子动力学模拟方法,进一步探索了Al熔体液态微观结构与熔体黏度的内在联系.含氢量的测定结果表明:氢含量随温度变化在780◦C发生突变.通过对液态Al的分子动力学模拟,发现Al的第一近邻配位原子的排布方式随温度的变化在780◦C左右与950◦C左右也存在突变.这似乎可以说明含氢量以及配位数的变化是导致黏度突变的原因.至于在升温和降温过程中发生黏度突变的温度不同,降温过程黏度突变点存在的滞后性,孙民华等认为是由于熔炼后的母合金存在微观不均匀性,升温过程消除了母合金的热历史所致.
为了更加具体地体现黏度在不同温度段的Arrhenius关系,将黏度取对数后,并对温度进行适当的变形,得到图4.可以看出,其黏度随温度的变化可以用三个Arrhenius公式来拟合.

图3 (a)加热时Al熔体的黏度随温度的变化;(b)降温时Al熔体的黏度随温度的变化[22]Fig.3.(a)Variation cure of Al melt viscosity with temperature during heating;(b)variation cure of Al melt viscosity with temperature during cooling[22].
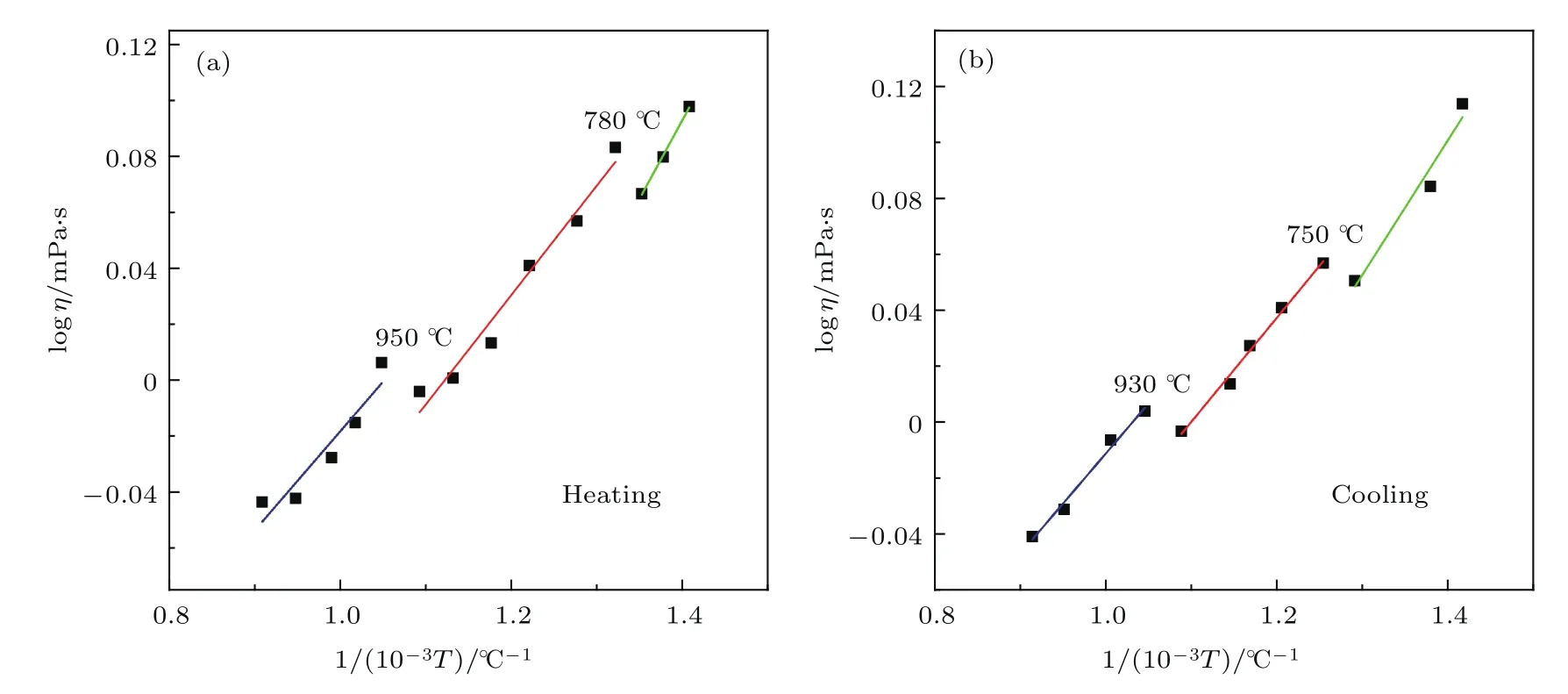
图4 (a)升温时Al熔体的Arrhenius曲线logη-1/(10−3T);(b)降温时Al熔体的Arrhenius曲线logη-1/(10−3T)Fig.4.(a)Arrhenius plot logη-1/(10−3T)of Al viscosity during heating;(b)arrhenius plot logη-1/(10−3T)of Al viscosity during cooling.
Wang等[23]报道了Sb,In,Sn,Bi高温金属熔体的黏度,并通过差示扫描量热法(DSC)扫描进行了分析.结果显示在黏度突变点附近,DSC曲线有明显的放热峰或吸热峰的存在,表明在黏度突变点附近高温金属熔体发生了热效应,此热效应是液态相变所致.Arrhenius曲线不连续点被认为是相变点,相变点把Arrhenius曲线分成几个不同的温度区域.在每一个温度区域范围内,公式中与黏度计算有关的系数η0和E不依懒于温度,只依赖于在某一特定的温度范围内液体的结构和弥散分布的流体团簇的尺寸.也就是说在黏度突变点前后的η0和E是不一样的,在没发生黏度突变之前这一温度范围内η0和E是一样的,不随温度的变化而变化.图5为Sb,In,Sn,Bi高温金属熔体的Arrhenius曲线,可以看出Sb,In的黏度突变现象并没有Sn,Bi那么明显,并且Sb,Sn,Bi只有一个黏度突变点,分别在670◦C,400◦C,480◦C发生突变,而In存在两个黏度突变点,在620◦C,300◦C发生突变.

图5 (a)Sb,(b)In,(c)Sn,(d)Bi Arrhenius曲线logη-1/(10−3T)[23]Fig.5.(a)Sb,(b)In,(c)Sn,(d)Bi Arrhenius plot logη-1/(10−3T)[23].
有关Pb[24−26],Ni[27−29],Ag[27,30,31]黏度,不同文献中报道的黏度值相差比较大.这极有可能是由于黏度的测量比较复杂、高温下熔体极易被氧化、不可控杂质浓度的影响、数据处理的方法或者实验条件的不同导致的.
3.2 二元合金体系的黏度
由于存在不同种类原子之间的相互作用,合金熔体的结构往往要比金属单质熔体结构复杂得多.合金在凝固的过程中除了存在能量起伏和结构起伏外,还存在着成分起伏.可以认为,合金有着比金属单质更为复杂的物化性质以及更广泛的应用领域.目前文献报道的二元合金体系的黏度值相对来说比金属单质要多.Zhao等[32]报道了不同组分下的CuZr二元合金体系,如图6所示.实验结果表明:不同组分下CuZr合金在1400 K左右均存在明显的黏度突变,说明液-液相变的发生;随着Cu原子分数的不断增加,黏度突变发生时对应的突变黏度值是不相同的,Cu60Zr40黏度突变时对应的突变黏度值最小;不同组分下CuZr合金发生黏度突变的温度范围不同,且Cu62Zr38黏度突变最缓慢(黏度突变在一个很宽的温度范围内完成).高温金属熔体在黏度突变范围以上被定义为高温转变区(HTZ),黏度突变范围以下被定义为低温转变区(LTZ).为了说明CuZr熔体的动力学变化,作者引用了过热熔体脆性的概念M,分别计算了HTZ和LTZ的M值,将MHTZ与MLTZ的比值定义为转变强度系数F.计算结果表明:MHTZ的值总是小于MLTZ的值,且F的值越大,玻璃形成能力越强.从这一结果可以看出,高温金属熔体黏度的变化情况极有可能成为衡量金属玻璃形成能力的一个重要判据.而这个工作也直接体现了金属玻璃制备过程中存在的液-固相关性.

图6 (a)Cu48Zr52,(b)Cu50Zr50,(c)Cu52Zr48,(d)Cu54Zr46,(e)Cu56Zr44,(f)Cu58Zr42,(g)Cu60Zr40,(h)Cu62Zr38合金Arrhenius曲线logη-1/(10−3T)[32]Fig.6.(a)Cu48Zr52,(b)Cu50Zr50,(c)Cu52Zr48,(d)Cu54Zr46,(e)Cu56Zr44,(f)Cu58Zr42,(g)Cu60Zr40,(h)Cu62Zr38melt Arrhenius polt logη-1/(10−3T)[32]
Zhao等[33]报道了Cu60Sn40黏度随温度变化的规律,并结合Cu-Sn合金相图分析了黏度非连续变化的原因.通过CuSn合金相图可以发现,化学计量数为Cu3Sn的成分在固态Cu-Sn合金中有一个很宽的存在范围,在CuSn高温金属熔体中,存在着Cu3Sn团簇结构.Cu60Sn40中的液-液相变,即黏度突变与Sn原子的聚集成键以及Cu3Sn团簇结构的形成有关.对于大多数物质,蒸发焓要比熔化焓大数倍,这就意味着Cu60Sn40熔体在刚开始熔化时只有部分的原子键破坏,剩余的大量原子以Cu3Sn团簇和Sn团簇的形式存在于高温金属熔体中.当温度超过900◦C时,Cu3Sn团簇中的Cu—Sn键以及Sn团簇中的Sn—Sn键被破坏,熔体倾向于均匀态,黏度也因此发生突变.同理在降温过程中,有相反的过程发生,Cu3Sn团簇和Sn团簇在900◦C附近生成,并随着温度的逐渐降低结合更加牢固,黏度再次出现突变.这一可逆过程可以表示为:L(Cu3Sn)+L(Sn)?L(Cu60Sn40).更多的研究结果证明了相图与高温金属熔体黏度突变可能存在的联系:有多种中间化合物生成的相图,其高温金属熔体的黏度突变程度往往比较明显[32,34];流体团簇的激活能的大小与液相线温度存在正相关性[35];在熔点或液相线温度以上相同的过热度条件下,有金属间化合物存在的区域黏度值较大[36].由于三元及以上相图比较复杂,目前高温合金熔体和相图的关系研究主要集中在二元合金.

图7 Cu60Sn40合金Arrhenius曲线logη-1/(10−3T)[33]Fig.7.Cu60Sn40melt Arrhenius plot logη-1/(10−3T)[33].
图8[15]是Ga80Cr20在不同磁场强度下黏度的Arrhenius曲线.在磁场强度分别为0,650,935 Gs时,Ga80Cr20均存在黏度突变,且黏度突变的温度范围一致.由图8可以看出,磁场强度在使黏度升高的同时不会使高温金属熔体黏度的突变行为消失.这说明黏度突变是金属玻璃高温金属熔体本质的体现,不随外界条件(比如磁场强度、升降温速过程)的改变而改变.Xu等[15]进一步定量描述了外加磁场与黏度之间的关系:黏度与磁场强度的平方成正比.二者之间的比例系数与电阻率有关,且随着熔体的温度的升高,电阻率逐渐增大,这样就可以通过对电阻率的测量来预测黏度在磁场作用下的变化情况.

图8 不同磁场强度下的Ga80Cr20Arrhenius曲线logη-1/(10−3T)[15]Fig.8.Ga80Cr20melt Arrhenius plot logη-1/(10−3T)in different magnetic field intensity[15].
Konstantinova等[37]在Cu-Al合金熔体的运动学黏度一文中,探究了Cu-Al合金熔体的运动学黏度随温度和成分的变化规律.图9所示为1100◦C CuAl合金随着Al含量的增加运动学黏度的变化情况.从图9可以看出在Cu70Al30附近运动学黏度有一个平缓的极值,在化学计量数为CuAl3附近运动学黏度有一个尖锐的极值.根据Arrhenius公式计算出的不同组分下的η0和E发现,在Cu70Al30附近有最大的E值,而在化学计量数为CuAl3附近有着最大η0值,这有可能是导致Cu-Al合金在图9相应的成分下出现极值的原因.

图9 Cu-Al合金体系等温运动学黏度[37]Fig.9.The isotherms of kinematic viscosity of melts of Cu-Al system[37].
3.3 多元合金体系的黏度
根据Inoue提出的制备块体非晶的三原则[38],我们已经知道制备大块非晶通常需具备的三个经验条件:1)合金由三种或以上组元组成;2)主要组元的混合焓为大的负值,即∆H<0;3)组元之间的原子尺寸比要大于12%.多元合金体系的黏度往往比单质或二元合金体系黏度值要大.Zhou等[34]在报道CuZr基熔体的液—液相变时,测量了Cu46Zr46Al8和Cu48Zr48Al4的黏度.实验结果表明,Cu46Zr46Al8和Cu48Zr48Al4均在1350 K左右存在明显的黏度突变,DSC扫描结果表明在1350 K左右有明显的放热峰存在,与黏度突变的温度区间较好地符合,两者均证明了液-液相变的发生.为了进一步探究黏度突变的动力学本质,他们用经验公式[23,39]计算了高温转变区和低温转变区流体团簇的尺寸,发现Cu46Zr46Al8在高温下团簇尺寸为7.62 nm,在低温下的团簇为6.12 nm;Cu48Zr48Al4在高温下团簇尺寸为9.58 nm,在低温下的团簇为7.69 nm.流体团簇的尺寸随着温度的降低,逐渐减小,通过流体团簇尺寸的变化说明,在降温的过程中,团簇结构变化是从高密度有序态变为低密度无序态的一个过程.
郭海东[19]报道了Co68.25Fe4.25Si15.25B12.25熔体的黏度[19],探究了Co-Fe-Si-B合金升温和降温过程中黏度值的差异.实验发现升温和降温过程中黏度数据有着明显不同,如图11所示.在升温过程中,Co-Fe-Si-B合金熔体是一个逐渐变化的过程,当温度达到1240◦C时,合金Co-Fe-Si-B熔体由低温结构转变为高温结构;在冷却过程中,当冷却到1240◦C时低温结构又再次恢复.这说明液态结构转变的可逆性.在降温过程中,其黏度值明显比升温过程同一温度下的黏度值低,这表明Co-Fe-Si-B熔体的高温结构具有一定的稳定性,结构转变的进行速度可能要比外界条件(如温度)的变化速度小得多.

图10 (a)降温过程中Cu48Zr48Al4(红色曲线)和Cu46Zr46Al8(黑色曲线)熔体的平衡黏度;(b)从1623 K以0.167 K/s速率降温的DSC曲线[34]Fig.10.(a)The equilibrium viscosity(η)upon cooling for Cu48Zr48Al4(red one)and Cu46Zr46Al8(black one)melts;(b)corresponding DSC outputs in arbitrary unit(AU)upon cooling from 1623 K with a 0.167 K/s rate[34].

图11 (a)Co68.25Fe4.25Si15.25B12.25熔体Arrhenius曲线logη-1/(10−3T);(b)升温和降温时Co68.25Fe4.25Si15.25B12.25熔体黏度变化曲线[19]Fig.11. (a)Co68.25Fe4.25Si15.25B12.25melt Arrhenius plot logη-1/(10−3T);(b)variation cure of viscosity of Co68.25Fe4.25Si15.25B12.25melt with temperature during heating or cooling[19].
铁基非晶的玻璃形成能力比较差,这就限制了铁基块体非晶的制备与应用.Zheng等[40]把铁基非晶低的玻璃形成能力归因于高温金属熔体的动力学转变(通过黏度的变化来体现),从流体团簇和过冷液体脆性角度出发,证实动力学行为的转折过程使铁基合金过冷液体变得非常不稳定,从而导致了铁基合金低的玻璃形成能力.此外研究还发现,动力学转变的程度对应着铁基合金不同的玻璃形成能力.如图12所示:A1,A2,A3分别代表着x=0,1,2时(Fe75B15Si10)100−xNbx(x=0,1,2)的组分,同样B1,B2,B3分别代表着x=0,1,2时Fe72−xCuxB20Si4Nb4(x=0,0.2,0.6)的组分. 从图12可以看出随着温度的降低黏度逐渐增大,然而斜率却发生了轻微的变化,黑色箭头所指的温度即为动力学转变的温度.分别计算了动力学转变前后HTZ和LTZ的η0和E,发现HTZ有较大的E值,但η0比LTZ要小,而LTZ有着较小的E值和大的η0.η0和E的变化证明了熔体动力学性质的变化.该变化的发生对应铁基熔体中高温有序态向低温无序态的变化.
关于多元合金体系黏度的报道还有很多,如Zhang等[41]报道了Al-Cu-Mg-Si体系黏度;Jia等[42]报道了Al-(Ni)-Yb体系黏度;Gancarz和Gasior[43]报道Sn-Zn-Na熔体的黏度;Liu等[44]报道了Fe-Ni-Co三元合金体系的黏度.这些研究大都集中在通过黏度突变探究高温金属熔体的液-液相变等方面,但是关于黏度突变的本质鲜有报道,液体结构仍然无法被很好解释.最近,Xiong等[45−50]通过实验和计算机模拟的手段研究了Al-Cu,Ag-Ge,Al-Ag,Ag-Ga合金原子结构的演变过程,实验结果是在熔点以上不同温度范围内通过高能X射线得到的结构因子S(q)曲线,通过观察S(q)曲线上峰值位置随温度的变化来观察液体结构的变化.研究发现,S(q)曲线上峰值发生变化的温度与G(r)曲线上峰值发生变化的温度符合得很好.实验和计算机模拟的手段同时证明了液体结构随温度的变化情况.

图12 黏度随温度的变化 (a)A系列,(b)B系列[40]Fig.12.Variation cure of viscosity of with temperature.(a)Series A;(b)series B[40].
4 高温金属熔体黏度研究的前景与展望
利用高温金属熔体的黏度变化趋势或规律,可以进一步获得过热熔体的脆性等动力学表征参数,同时也是研究液-液相变现象、金属玻璃形成液体的强脆转变现象的重要依据.作为一种由高温金属熔体经快速冷却凝固制备而成的先进材料,金属玻璃的制备更容易受到高温金属熔体黏度的直接影响.理解合金高温金属熔体的动力学性质变化规律,不仅有助于从液-固遗传的角度理解金属凝固过程的本质、金属晶化与非晶化的竞争关系以及金属材料固体性质与其液体动力学脆性的联系,同时对生产实践也具有重要的指导意义.如果把超快速凝固也作为一种先进铸造技术来看待,液-固遗传性必然在金属玻璃形成体系中同样存在.而且,由于金属玻璃的整体结构更接近液体的结构,因此相比较传统铸造技术制备的金属材料,金属玻璃高温金属熔体的性质应该对其固体的性质产生更为直接的影响.前期工作初步发现了高温金属熔体的黏度异常突变与非晶固体三阶段焓异常弛豫行为的关系[51,52].具有明显三阶段焓异常弛豫行为的非晶合金,其高温金属熔体的黏度突变也非常明显.而高温金属熔体黏度突变不明显的玻璃形成体系,其固体焓弛豫行为具有单调性,仅仅在非晶条带的起始弛豫激活能上可以观察到三阶段的变化.高温金属熔体的黏度突变程度还与玻璃形成能力有关.已经发现,金属过热液体的脆性与玻璃形成能力具有反比关系.进一步地,在发生液-液相变的金属玻璃熔体体系中,其发生液-液相变的相变程度也与玻璃形成能力密切相关[32,40],但这种关系似乎更为复杂.在CuZr体系中,发生的黏度突变对应明显的放热过程[32],而在铁基玻璃形成体系中,其黏度的突变伴随微弱的吸热效应[40].这一比较似乎可以说明发生液-液相变后的CuZr熔体结构更为稳定,而铁基则刚好相反.高温金属熔体黏度的异常变化同样可以引起固体力学性质和其他物理性质的变化.通过对26种不同种类液体的数据分析,金属玻璃液体的强脆转变系数(即黏度随温度的变化率的改变)与非晶固体的泊松比成负的指数关系[53,54].目前,对金属玻璃熔体的认识还不够深入,对其黏度突变伴随的微观结构以及其他物性的变化还不明确.对于高温金属熔体黏度与液态金属键的关系,国内外研究较少.键型数的改变与黏度的改变有无因果关系,团簇的哪些特征对黏度突变产生明显的影响,仍缺乏定量与定性的分析.飞速发展的中子散射技术、高能X射线衍射技术等先进实验手段的应用,辅助以计算机模拟,以原子径向密度分布函数、双体分布函数、均方位移等多种结构参数为桥梁,结合应用键对分析技术等,使深入探索高温金属熔体黏度突变的微观结构来源成为可能.
另外,作为体现材料在不同温度、压力、成分下组织和结构变化的依据,相图可以直观体现不同成分下的合金在不同温度范围内所处的组织状态.高温金属熔体的黏度变化是由于液体结构变化所致,将相图与高温金属熔体的黏度结合在一起,或许是探究高温金属熔体的黏度突变或液-液相变本质的另一条出路[32,34−36].相图与高温金属熔体黏度之间的内在关系尚未明确,还需要进行更为系统和深入的研究.以二元合金成分与黏度之间的关系为例,MolewynHoghes提出了一种理论[55],其表达式为
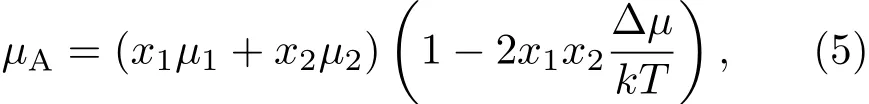
式中µA为合金的运动学黏度;x1,x2分别为两种成分的摩尔分数;∆µ为转换能;µ1,µ2分别为两种成分的运动学黏度.但用该理论计算的结果与实验结果相比较,存在着较大误差.桂满昌[56]通过对Al-Si合金的黏度研究发现:在共晶点左边亚共晶区域内,随着含硅量的增加,黏度值下降;在共晶点右边的过共晶区域,随着含硅量的增加,黏度值增高.此外,在亚共晶铝硅合金中,加入变质元素Sr使合金的黏度增加,同时使黏度突变量减小,但并不改变黏度突变点的温度;在过共晶铝硅合金中加入变质元素P后,使合金第一个黏度突变点温度升高,并使合金黏度值增加.除此之外,其他学者也提出了一些有关二元合金成分与黏度之间关系的理论或半经验的模型[57−61].但是这些模型往往只适用于某些特定的二元合金体系,当换用其他体系时,存在着比较大的误差.由此可见黏度与高温金属熔体成分之间的关系比较复杂,对其关系的确立必须建立在大量准确的黏度数据基础之上.
[1]Han X F 2005M.S.Dissertation(Jinan:Shandong University)(in Chinese)[韩秀峰 2005硕士学位论文 (济南:山东大学)]
[2]Angell C A 1985J.Non-Cryst.Solids73 1
[3]Bian X F,Sun B A,Hu L N,Jia Y B 2005Phys.Lett.A335 61
[4]Meng Q G,Zhou J K,Zheng H X,Li J G 2006Scr.Mater.54 777
[5]Hu L N,Bian X F 2003Chin.Sci.Bull.48 2393(in Chinese)[胡丽娜,边秀房 2003科学通报 48 2393]
[6]Hu L N,Zhang C Z,Yue Y Z,Bian X F 2010Chin.Sci.Bull.55 115(in Chinese)[胡丽娜,张春芝,岳远征,边秀房2010科学通报55 115]
[7]Books R F,Dinsdale A T,Quested P N 2005Meas.Sci.Technol.16 354
[8]Dinsdale A T,Quested P N 2004J.Mater.Sci.39 7221
[9]Torklep K,Oye H A 1979J.Phys.E12 875
[10]SatoY,KamedaY,NagasawaT,SakamotoT,Moriguchi S,Yamamura T,Waseda Y 2003J.Cryst.Growth249 404
[11]Kehr M,Hoyer W,Egry I 2007Int.J.Thermophys.28 1017
[12]Nunes V M B,Santos F J V,de Castro C A N 1998Int.J.Thermophys.19 427
[13]Schenck H,Frohberg M G,Hoffmann K 1963Steel Res.Int.34 93
[14]Emadi D,Gruzleski J E,Toguri J M 1993Metall.Trans.B24 1055
[15]Xu Y P,Zhao X L,Yan T L 2017Chin.Phys.B26 036601
[16]Wu Y Q,Bian X F,Mao T,Li X L,Li T B,Wang C D 2006Phys.Lett.A361 265
[17]Sun C,Geng H,Liu J,Gneg H,Yang Z 2004Phys.Meas.1 16
[18]Wang C Z 2017M.S.Dissertation(Jinan:Shandong University)(in Chinese)[王春震 2017硕士学位论文 (济南:山东大学)]
[19]Guo H D 2008M.S.Dissertation(Harbin:Harbin Institute of Technology)(in Chinese)[郭海东 2008硕士学位论文(哈尔滨:哈尔滨工业大学)]
[20]Sun C J,Geng H R,Zhang N,Teng X Y,Ji L L 2008Mater.Lett.62 73
[21]Mao T,Bian X F,Morioka S,Wu Y Q,Li X L,Lü X Q 2007Phys.Lett.366 155
[22]Sun M H,Geng H R,Bian X F,Liu Y 2000Acta Metal.Sin.36 1134(in Chinese)[孙民华,耿浩然,边秀房,刘燕2000金属学报36 1134]
[23]Wang L,Bian X F,Liu J T 2004Phys.Lett.A326 429
[24]Ofte D,Wittenberg L J 1963Trans.Metall.Soc.Aime.227 706
[25]Rothwell E 1961J.Inst.Metals90 389
[26]Gebhardt E,Kostlin K 1957Z.Metallkd.48 636
[27]Schenck H,Frohberg M G,Hoffmann K 1963Arch.Eisenhuettenw.34 93
[28]Cavalier G 1963Compt.Rend.256 1308
[29]Kaplun A B,Avaliani M 1977High Temp.15 259
[30]Nikolaev B,Vollmann J 1996J.Non-Cryst.Solids208 145
[31]Martin-Garin L,Martin-Garin R,Despre P 1978J.Less Common.Met.59 1
[32]Zhao X,Wang C Z,Zheng H J,Tian Z A,Hu L N 2017Phys.Chem.Chem.Phys.19 15962
[33]Zhao Y,Hou X X,Bian X F 2008Mater.Lett.62 3542
[34]Zhou C,Hu L N,Sun Q J,Bian X F,Yue Y Z 2013Appl.Phys.103 171904
[35]Ning S,Bian X F,Ren Z F 2010Phys.B:Condens.Matter405 3633
[36]Mao T,Bian X F,Xue X Y,Zhang Y N,Guo J,Sun B A 2007Phys.B:Phys.Condens.Matter387 1
[37]Konstantinova N Y,Popel’P S,Yagodin D A 2009High Temp.47 336
[38]Inoue A,Takeuchi A 2010Int.J.Appl.Glass Sci.1 273
[39]Wang L,Liu J T 2004Phys.Lett.A328 241
[40]Zheng H J,Hu L N,Zhao X,Wang C Z,Sun Q J,Wang T,Hui X D,Yue Y Z,Bian X F 2017J.Non-Cryst.Solids471 120
[41]Zhang F,Du Y,Liu S H,Jie W Q 2015Comput.Coupling Phase Diagrams Thermochem.49 79
[42]Jia R,Bian X F,Lu X Q,Song K K,Li X L 2010Phys.Mech.Astron.53 390
[43]Gancarz T,Gasior W 2016Fluid Phase Equilib.418 57
[44]Liu Y H,Lu X W,Bai C G,Lai P S,Wang J S 2015J.Ind.Eng.Chem.30 106
[45]Xiong L H,Lou H B,Wang X D,Debela T T,Cao Q P,Zhang D X,Wang S Y,Wang C Z,Jiang J Z 2014Acta Mater.68 1
[46]Xiong L H,Chen K,Ke F S,Lou H B,Yue G Q,Shen B,Dong F,Wang S Y,Chen L Y,Wang C Z,Ho K M,Wang X D,Lai L H,Xiao T Q,Jiang J Z 2015Acta Mater.92 109
[47]Xiong L H,Yoo H,Lou H B,Wang X D,Cao Q P,Zhang D X,Cao Q P,Zhang D X,Jian J Z,Xie H L,Xiao T Q,Jeon S,Lee G M 2015J.Phys.:Condens.Matter27 035102
[48]Xiong L H,Guo F M,Wang X D,Cao Q P,Zhang D X,Ren Y,Jiang J Z 2017J.Non-Cryst.Solids459 160
[49]Xiong L H,Wang X D,Cao Q P,Zhang D X,Xie H L,Xiao T Q,Jiang J Z 2017J.Phys.:Condens.Matter29 035101
[50]Su Y,Wang X D,Yu Q,Cao Q P,Ruett U,Zhang D X,Jiang J Z 2018J.Phys.:Condens.Matter30 015402
[51]Wang C W,Hu L N,Chen W,Tong X,Zhou C,Sun Q J,Hui X D,Yue Y Z 2014J.Phys.Chem.141 164507
[52]Hu L N,Zhou C,Zhang C Z,Yue Y Z 2013J.Phys.Chem.138 174508
[53]Sun Q J,Hu L N,Zhou C,Zheng H J,Yue Y Z 2015J.Phys.Chem.Lett.143 164504
[54]Sun Q J,Zhou C,Yue Y Z,Hu L N 2014J.Phys.Chem.Lett.5 1170
[55]Iida T,Roderick I L,1993The Properties of Liquid Metals(Oxford:University Press)pp147–199
[56]Gui M C 1994Ph.D.Dissertation(Harbin:Harbin Institute of Technology)(in Chinese)[桂满昌 1994博士学位论文(哈尔滨:哈尔滨工业大学)]
[57]Iidia T,Ueda M,Morita Z 1976Tetsu to Hagane62 1169
[58]Morita Z,Iida T,Ueda M 1997Inst.Phys.Conf.Ser.30 600
[59]Djemili B,Martin-Garin L,Hicter P 1980J.Phys.Colloq.C841 363
[60]Enskog D 1922Arkiv.Mth.Astron.Fys.16 16
[61]Tham M K,Gubbins K E 1971J.Chem.Phys.55 268
猜你喜欢
作文周刊·小学一年级版(2022年24期)2022-06-18 13:11:03
中学生数理化·八年级物理人教版(2022年4期)2022-04-26 14:11:14
环境保护与循环经济(2021年7期)2021-11-02 08:10:52
食品安全导刊(2021年20期)2021-08-30 06:39:22
中学生数理化·八年级物理人教版(2021年4期)2021-07-22 03:15:16
小天使·一年级语数英综合(2017年1期)2017-02-16 10:17:13
中国塑料(2016年4期)2016-06-27 06:33:48
中国塑料(2016年3期)2016-06-15 20:30:01
华东理工大学学报(自然科学版)(2015年3期)2015-11-07 09:17:13
华东理工大学学报(自然科学版)(2015年3期)2015-11-07 09:17:13
