
RNA质量对内参基因选择和qRT-PCR结果评价的影响
2018-06-13 03:34:22霍雪萍张琳萍赵向绒刘勤社王海芳
生物技术通讯 2018年3期
霍雪萍,张琳萍,赵向绒,刘勤社,王海芳
1.陕西省人民医院 中心实验室,陕西 西安 710068;2.西安交通大学 医学部中西医结合专业,陕西 西安 710061;3.陕西省中西医结合心血管病防治重点实验室,陕西中医药大学,陕西 西安 712046
在研究分子机理时,检测相关基因在mRNA水平的表达差异是重要环节,目前多采用实时定量 荧 光 PCR(quantitativereal-timePCR,qRTPCR)方法,具有定量准确、灵敏度高、快速简便及高通量等特点[1]。制备高质量的RNA,选定表达稳定的内参基因,对目的基因的相对表达量进行校正,是获得真实准确的实验数据的前提。
核酸质量的控制是对qRT-PCR实验结果质量控制的第一步。核酸在260nm处有最大吸收峰,蛋白质在280nm处有最大吸收峰,盐和小分子在230nm处有最大吸收峰。D260nm/D280nm和D260nm/D230nm是核酸纯度的指示。高质量RNA的D260nm/D280nm能达到 1.8~2.2,如果比值低于 1.8,表示存在蛋白质或酚类物质的影响;D260nm/D230nm值应大于2.0,若比值小于2.0则表明样品被糖类、盐类或有机溶剂污染[2]。以往的研究关注D260nm/D280nm值对qRT-PCR结果的影响较多,而对D260nm/D230nm值的影响关注较少。
我们在利用qRT-PCR法检测肿瘤坏死因子α(tumornecrosisfactor α,TNFα)调控下游细胞间黏附分子1(intercellularcelladhesionmolecule-1,ICAM-1)基因表达的研究中,发现RNA质量对于内参基因的选择和基因表达水平的评价分析具有显著影响。在此进行报告,以期为相关研究提供参考。
1 材料与方法
1.1 材料
小鼠动脉内皮细胞(arterialendothelialcell,AEC)购自上海拜力生物科技有限公司;RPMI-1640细胞培养基和胎牛血清购自Clontech公司;细胞培养用青霉素/链霉素和胰蛋白酶购自碧云天生物技术研究所;重组小鼠TNFα购自北京义翘神州生物技术公司;总RNA提取试剂盒购自北京天根生化科技有限公司。
1.2 细胞培养及药物处理
小鼠AEC贴壁生长于含10%胎牛血清和1%双抗的RPMI-1640培养液中,在37℃、5%CO2及饱和湿度的恒温密闭细胞培养箱内培养,0.025%胰蛋白酶消化传代,取对数生长期细胞用于实验。将6孔细胞培养板中长满(80%~90%)的AEC分为正常组和TNFα组。A批次的TNFα组为50 ng/mLTNFα诱导6h,B批次的TNFα组为30ng/mLTNFα诱导6h。
1.3 总RNA提取及cDNA合成
收集细胞,加入1mLTRIzol试剂,按说明书实验流程提取细胞总RNA,用NanoDrop2000C紫外分光光度计(Thermoscientific公司)检测总RNA浓度和纯度,每个样品上样量为1μL。D260nm值作为RNA浓度测定的指标(以ng/mL为单位),D260nm/D280nm和D260nm/D230nm比值作为RNA纯度的指标。反转录反应采用PrimeScriptRTMaster Mix(PerfectRealTime)试剂盒(TaKaRa公司)。反转录体系为10μL,以500ng总RNA为起始材料,5×PrimeScriptRTMasterMix2 μL,无 RNA酶水补足10μL。反转录条件:37℃ 15min,85℃ 5s,4℃保存。
1.4 引物设计与合成
用NCBI软件设计β-actin、GAPDH和ICAM-1引物,由上海生工生物工程股份有限公司合成,引物信息见表1。
1.5 qRT-PCR法检测基因表达
用 SYBR Premix ExTaqⅡ(Tli RNaseH Plus)试剂盒(TaKaRa公司)和ABI7500荧光PCR扩增仪(Life公司)进行荧光定量PCR检测。PCR反应体系:SYBR Premix ExTaqⅡ(TilR NaseH Plus)10 μL,模板 cDNA1 μL,正反引物各 0.5 μL,ddH2O8μL,总体系20μL。所有操作在冰上完成,每个样品设立3个复孔。反应条件:95℃预变性 5min,95℃变性 1min,58℃退火 45s,72℃延伸45s,在延伸阶段进行荧光信号采集,40个循环。分别以β-actin、GAPDH为内参基因,计算同一样本中目的基因与内参基因的含量比值,评价目的基因表达水平。

表1 引物信息
1.6 统计学方法
从ABI7500荧光定量PCR仪中导出阈值循环数(Ct),换算各基因的相对表达量为 2-ΔΔct,采用Excel软件进行统计学分析。
2 结果
2.1 RNA质量评价
用NanoDrop2000C紫外分光光度计对RNA的浓度和纯度进行检测。A组样品RNA的D260nm/D280nm为 1.8~2.2,D260nm/D230nm>2.0,表 明 该 组 样 品RNA纯度好;B组样品RNA的D260nm/D280nm为1.8~2.2,D260nm/D230nm<2.0,表明该组样品可能被碳水化合物(糖类)、盐类或有机溶剂污染。见表2。
2.2 qRT-PCR基因检测结果
以1μLcDNA作为qRT-PCR的起始模板,确保cDNA大致相同的拷贝数,以保证后续结果具有可比性。图1为内参基因和目标基因的溶解曲线,出现单一的信号峰,说明没有引物二聚体和非特异性条带产生,引物设计合理,结果准确可靠,满足实时荧光定量PCR对引物的要求。表3为A、B批次样品PCR反应中2种内参基因及目的基因ICAM-1的mRNA表达水平。A批次样品β-actin和GAPDH基因的Ct值小于20,2个内参基因的表达量均较高且一致性好;B批次样品β-actin基因的Ct值大于20,GAPDH基因的Ct值小于20,组内一致性均较好。以上数据表明实验操作过程无误,数据可信度高。
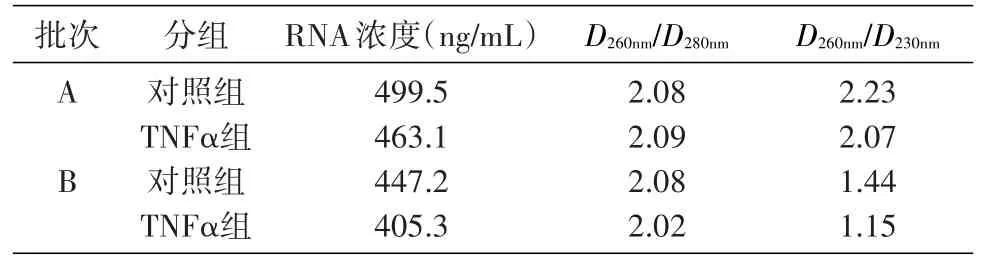
表2 RNA质量对照表
2.3 qRT-PCR结果分析
采用 2-ΔΔct法对 ICAM-1 的 mRNA 水平进行相对定量分析。A批次,采用2种内参基因进行相对定量,TNFα组的ICAM-1mRNA表达水平非常接近;B批次,以β-actin和以GAPDH作为内参基因对ICAM-1相对定量,TNFα组的表达水平差别近30倍(表4)。
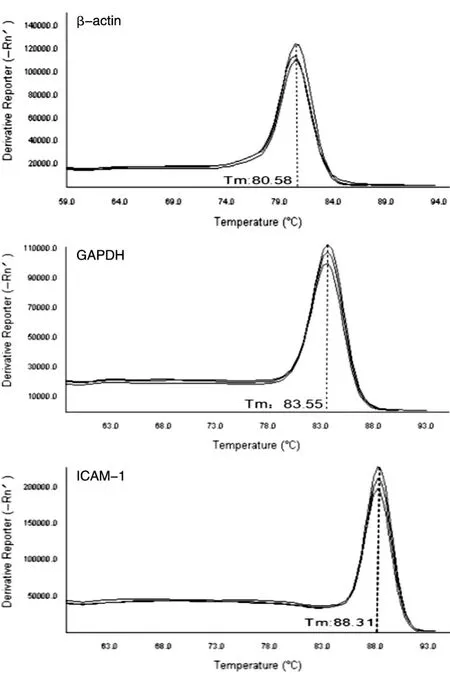
图1 基因熔解曲线
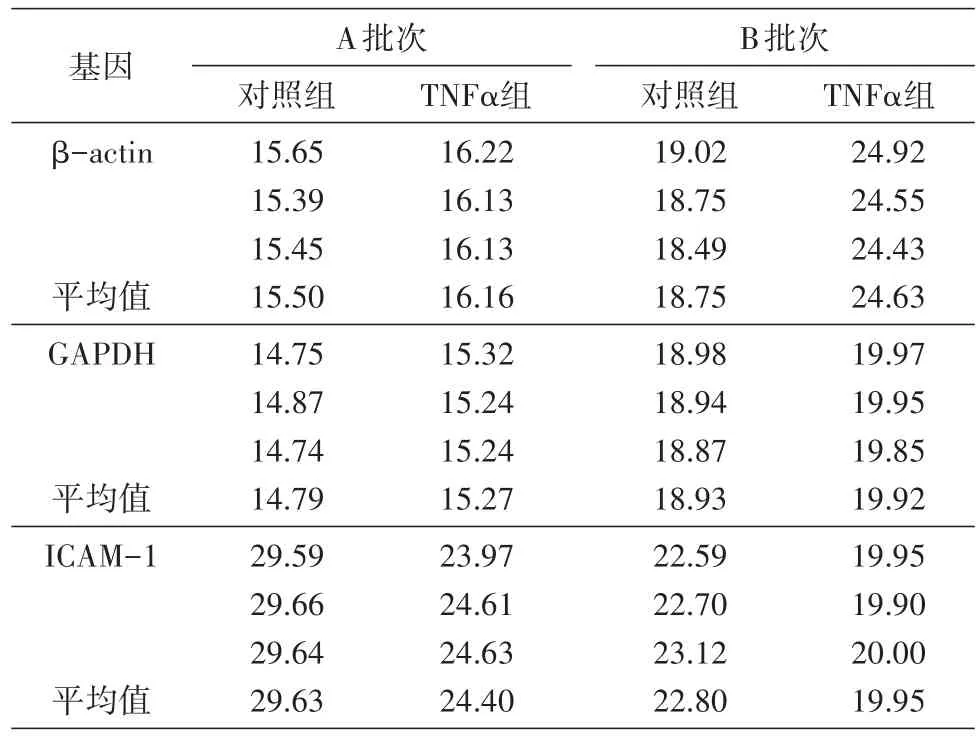
表3 qRT-PCR基因检测结果(Ct值)
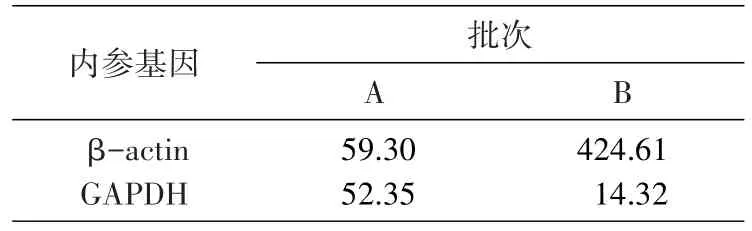
表4 内参基因对目标基因表达结果分析的影响
3 讨论
实时荧光定量PCR是分析基因表达的一种有效方法,但分析结果会受RNA质量、RNA反转录、扩增效率等多种因素的影响[3]。为了避免这类误差的产生,在分析目标基因的表达量时,须选择表达稳定性较高的内参基因作为校正标准。然而,任何一种内参基因的所谓稳定表达都是在一定类型的细胞或实验因素作用下在一定范围内的恒定,并非绝对的稳定表达[4]。许多研究证实理想的内参基因并不存在,不同的处理因素以及不同组织中内参基因表达的稳定性是不一致的[5-6]。因此,在利用qRT-PCR进行基因表达的研究时,应根据不同的实验条件、实验样品选用合适的内参基因。
TNFα是机体急、慢性炎症反应的重要细胞因子,能够诱导多种黏附分子的表达,促进单核细胞浸润,参与炎症病理进程。ICAM-1是公认的血管炎性疾病标志物之一。我们发现在研究TNFα作用的文献中,有关ICAM-1分子水平的检测大多采用β-actin作为内参基因。我们采用2种常用内参基因GAPDH和β-actin进行研究时发现,RNA质量对于内参基因的选择和qRT-PCR结果评价具有重要意义。RNA的D260nm/D280nm和D260nm/D230nm比值都应当加以考虑。当核酸D260nm/D280nm在正常范围内,D260nm/D230nm>2.0,即RNA纯度很高时,采用2种内参对于目的基因ICAM-1的相对表达量评价结果相近;如果D260nm/D230nm<2.0,即RNA纯度不高时,采用不同内参对于ICAM-1基因的相对表达量评估存在很大差异,比较而言,此时选用GAPDH内参评价结果的可信度更高。因此,在进行TNFα的相关分子研究以及对珍稀样品进行生物学研究时,应针对核酸质量选择合适的内参基因进行qRT-PCR实验,必要情况下可选择至少2个内参,综合分析,得出可靠的实验结果。
猜你喜欢
粉末冶金技术(2021年3期)2021-07-28 06:26:50
世界科学技术-中医药现代化(2020年2期)2020-07-25 02:06:06
传媒评论(2019年12期)2019-08-24 07:55:10
中成药(2018年12期)2018-12-29 12:25:44
童话世界(2017年29期)2017-12-16 07:59:32
传媒评论(2017年3期)2017-06-13 09:18:10
中成药(2017年6期)2017-06-13 07:30:35
中学生数理化·高二版(2016年6期)2016-05-14 13:19:33
新闻传播(2015年10期)2015-07-18 11:05:39
医学研究杂志(2015年4期)2015-06-10 06:42:43
