
7-卤代吲哚的合成工艺研究
2018-06-09 07:35:14刘长春蒋若愚刘承先李雪莲薛叙明
精细石油化工 2018年3期
刘长春,程 进,蒋若愚,刘承先,李雪莲,薛叙明
(常州工程职业技术学院化学与材料工程学院,江苏 常州 213164)
7-卤代吲哚是一类重要的吲哚化合物,具有独特的化学结构、较强的生物活性和药理活性,广泛应用于医药、农药以及有机合成领域。7-卤代吲哚可用于合成治疗糖尿病高血糖症的钠依赖性葡萄糖共转运蛋白2抑制剂[1]、治疗关节炎的β-胰蛋白酶抑制剂[2-3]、治疗肥胖症的蛋白偶联受体GPR119激动剂[4]、治疗神经性疼痛的大麻素CB1受体激动剂[5-6]等多种药物[7-13]和杀菌剂[14, 15]等。
合成吲哚骨架的反应有很多,但制备7-卤代吲哚的反应相对较少。目前报道的制备7-卤代吲哚的方法主要有:1)Bartoli吲哚合成法,2-卤代硝基苯与三倍量乙烯基格式试剂在-78 ℃直接反应制备7-卤代吲哚[16-18],该法反应温度极低,条件苛刻;2)Leimgruber-Batcho吲哚合成法,以3-卤代邻硝基甲苯与DMF-DMA经缩合、还原及分子内关环反应制备7-卤代吲哚,但原料3-卤代邻硝基甲苯相对较贵[19-20];3)吲哚为原料制备法,吲哚与甲酸及1-(3-二甲基氨基丙基)-3-乙基碳二亚胺经酰基化、铊化、卤代、碱水解、脱氢反应后制备得7-卤代吲哚[21-22],但铊化合物剧毒,且反应步骤过于繁琐,收率低;4)Sugasawa合成法,苯胺与氯乙腈经佛克烷基化、还原关环合成7-卤代代吲哚[23-24],但氯乙腈极毒危险。另外还有通过Stille偶联反应[25]、Sonogashira偶联[26-28]、微波反应法[29]等法制备7-卤代吲哚,但普遍使用剧毒品作为原料,工艺路线较复杂,不宜推广。
本文以邻卤代苯胺和水合氯醛、盐酸羟胺为原料,通过Sandmeyer异亚硝基乙酰替苯胺合成法制备得7-卤代靛红,再经硼氢化钠/三氟化硼乙醚体系还原制备得7-卤代吲哚,合成路线如图1所示。由价格相对较低的靛红还原制备吲哚的工艺,目前尚未见报道。该工艺具有原料成本低,反应条件温和,收率较高,操作简便等优点,适合批量制备7-卤代吲哚,具有较好的工业应用前景。
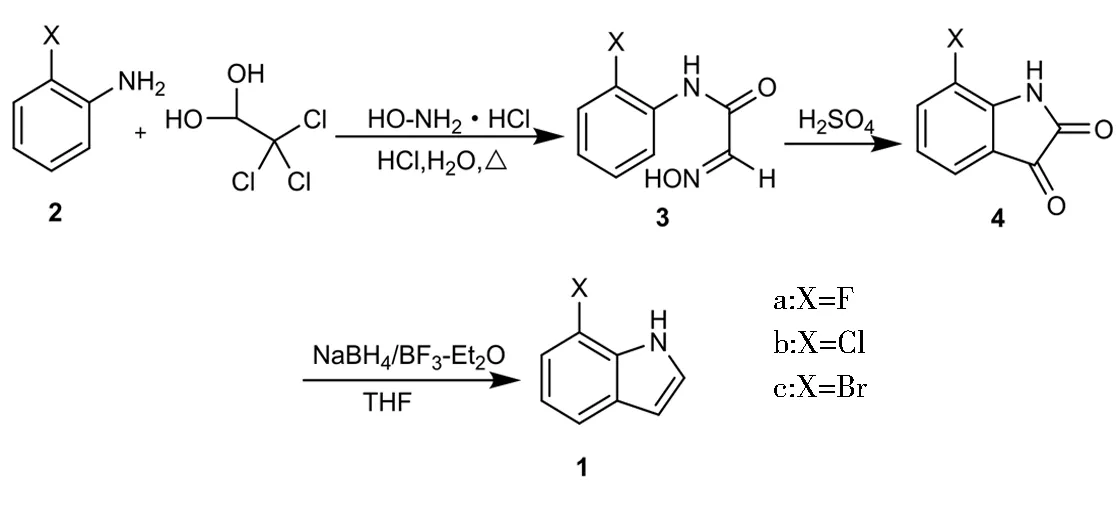
图1 吲哚合成路线
1 实验部分
1.1 主要试剂及仪器
邻氟苯胺、邻氯苯胺、邻溴苯胺、硼氢化钠,以上均为分析纯,国药集团化学试剂有限公司;浓盐酸、无水硫酸钠、水合氯醛、浓硫酸、四氢呋喃、硫酸氢钠、乙酸乙酯、无水硫酸镁、异丙醇,以上均为化学纯,江苏强盛功能化学有限公司;三氟化硼乙醚,乙硼烷四氢呋喃溶液,以上均为分析纯,阿拉丁试剂有限公司。
Mercuryplus 400 MHz型核磁共振仪,瑞士Bruker公司;Waters515高效液相色谱仪,美国WATERS公司;Elementar Vario 400元素分析仪,瑞士Bruker公司。
1.2 实验步骤
1.2.1N-(2-卤苯基)-2-异亚硝基乙酰苯胺(3a,3b,3c)的合成
向100 mL带搅拌的四口烧瓶中,依次加入25 mL水,10 mL(0.12 mol)浓盐酸, 0.09 mol邻卤代苯胺(10.00 g 2a或11.48 g 2b或15.48 g 2c)制得邻卤代苯胺盐酸盐备用。在另一250 mL四口烧瓶中加入100 mL水,71.60 g(0.50 mol)无水硫酸钠,搅拌升温至35 ℃溶解后,加入16.50 g(0.10 mol)水合氯醛,滴加入上述邻卤代苯胺盐酸盐,最后加入20.00 g(0.29 mol)盐酸羟胺。加料毕,搅拌加热反应(制备3a时约回流反应10 min;制备3b时80 ℃反应约4 h;制备3c时80 ℃反应约3 h ),薄层色谱(TLC)监控。反应毕,反应液冷却至0 ℃后,过滤,洗涤,干燥,化合物3a~3c。酸性滤液在下一批次投料中替代水循环套用。
3a:15.97 g黄褐色粉末状固体N-(2-氟苯基)-2-异亚硝基乙酰苯胺,收率97.42%,含量98.34%,熔点118~119 ℃(文献[30]: 120 ℃)。1H NMR(400 MHz,DMSO),δ:7.08(m,1H,Ar—H),7.17(m,1H,Ar—H),7.29(m,1H,Ar—H),7.89(m,1H,Ar—H),8.29(s,1H,C—H),9.87(s,1H,N—H),12.01(s,1H,O—H)。13CNMR(100 MHz, CDCl3),δ:115.1,123.5,128.5,130.7,143.9,146.3,157.6,161.3。元素分析,C8H7FN2O2,实测值(计算值),%:C 52.68 (52.75);H 3.89(3.87)。
3b:12.58 g黄色粉末状固体N-(2-氯苯基)-2-异亚硝基乙酰苯胺,收率70.37%,含量98.44%,熔点150~151 ℃(文献[33]:151 ℃)。1H NMR(DMSO,400 MHz),δ:7.12(m,1H,Ar—H),7.28(m,1H,Ar—H),7.59(m,1H,Ar—H),8.01(m,1H,Ar—H),8.26(s,1H,C—H),9.85(s,1H,N—H),11.11(s,1H,O—H)。13C NMR(100 MHz,CDCl3),δ:123.1,124.8,128.7,130.0,131.5,135.2,147.1,161.4。元素分析,C8H7ClN2O2,实测值(计算值),%:C 49.11(48.38);H 2.57(3.55)。
3c:18.78g黄色粉末状固体N-(2-溴苯基)-2-异亚硝基乙酰苯胺,收率85.83%,含量98.47%,熔点167~169 ℃(文献[36]:170 ℃)。1H NMR(DMSO, 400 MHz),δ:7.18(m,1H,Ar—H),7.46(m,1H,Ar—H),7.90(m,1H,2*Ar—H),8.31(s,1H,C—H),9.87(s,1H,N—H),12.41(s,1H,O—H)。13C NMR(100 MHz, CDCl3),δ:116.9,125.0,127.5,132.3,135.6,146.7,161.3。元素分析,C8H7BrN2O2,实测值(计算值),%:C 39.51 (39.53);H 2.89(2.90)。
1.2.27-卤代靛红(4a,4b,4c)的合成
在250 mL四口烧瓶中,将0.05 molN-(2-卤代苯基)-2-异亚硝基乙酰苯胺(9.11 g 3a或9.93 g 3b或12.15 g 3c)分批加入至20mL浓硫酸中,加料过程温度控制在60~70 ℃,加料毕,升温至80~85 ℃搅拌反应(制备4a、4c时80 ℃;制备4b时85 ℃),TLC监控,反应毕,在剧烈搅拌下将反应液倒入200 g碎冰中淬灭,搅拌1 h后,过滤、干燥得7-卤代靛红(4a~4c)。
4a:8.14 g黄色固体7-氟靛红,收率98.65%,含量98.93%,熔点194~196℃(文献[31]:194 ℃)。1H NMR(400 MHz,DMSO),δ:7.04(m,1H,Ar—H),7.28(t,J=7.4,1H,Ar—H),7.79(d,J=10.5,1H,4—H),11.23(s,1H,N—H)。13C NMR(100 MHz,DMSO), δ:118.4,120.8,125.9,132.4,158.9,164.6,183.7。元素分析,C8H4FNO2,实测值(计算值),%:C 58.11 (58.19);H 2.46(2.44)。
4b:8.46 g淡黄色固体7-氯靛红,收率93.49%,含量98.64%,熔点186~187℃(文献值[34]: 184~186 ℃)。1H NMR(DMSO, 400 MHz),δ:7.44(m,1H,Ar—H),7.80(m,2H,Ar—H), 11.26(brs,1H,N—H)。13C NMR(100 MHz, CDCl3),δ:119.0,123.4,130.6,132.8, 135.5,134.3,159.5, 184.3。元素分析,C8H4ClNO2,实测值(计算值),%:C 52.90 (52.92);H 2.23(2.22)。
4c:9.94g黄色固体7-溴靛红,收率88.37%,含量97.91%,熔点194~195 ℃(文献值[37]192~194 ℃)。1H NMR(DMSO, 400 MHz),δ:7.02(t,J=7.5 Hz,1H, Ar—H),7.51(d,J=7.2 Hz,1H,Ar—H),7.79(q,J=8.1 Hz,1H,Ar—H),11.33(s,1H,N—H)。13C NMR(100 MHz, CDCl3),δ:119.1,122.8,124.6,126.1,137.4,143.6,160.6,182.3。元素分析,C8H4BrNO2,实测值(计算值),%:C 42.51(42.51);H 1.79(1.78)。
1.2.37-卤代吲哚(1a,1b,1c)的合成
将0.024 mol 7-卤代靛红(3.96 g 4a或4.34 g 4b或5.40 g 4c)和2.72 g(0.072 mol)硼氢化钠置于一干燥四口烧瓶中,冷却至-10 ℃以下,搅拌下向其中缓慢滴加40 mL干燥的四氢呋喃和7.66 g(0.054 mol)三氟化硼乙醚,整个过程控制温度不超过-5 ℃,TLC监控,反应10 h,反应毕,向其中缓慢滴入6.60 g硫酸氢钠和70 mL水组成的水溶液淬灭(注意防止爆沸),混合液经乙酸乙酯萃取,无水硫酸镁干燥,减压蒸除溶剂,异丙醇重结晶后,得7-卤代吲哚(1a~1c)。
1a:2.58 g白色针状固体7-氟吲哚,收率79.55%,含量99.51%,熔点60~62 ℃(文献值[32]61~62℃)。1H NMR (400 MHz, CDCl3),δ:6.58(d,J=3.6,1H,3-H),6.90(m,1H,2-H),7.02(m,1H,Ar—H),7.23(q,J=15.8,1H,Ar—H),7.40(d,J=13.1,1H,Ar—H),8.36(br s,1H,N—H)。13C NMR(100 MHz, CDCl3),δ:102.2,108.9,116.2,121.8,124.0,128.9,130.8,149.6。元素分析,C8H6FN,实测值(计算值),%:C 71.13 (71.10);H 4.53(4.48)。
1b:2.69 g白色针状固体7-氯吲哚,收率73.96%,含量99.24% ,熔点57~58 ℃(文献值[35]58 ℃)。1H NMR(DMSO, 400 MHz),δ:6.56(dd,J=2.7Hz,C—H),7.03(m,1H,Ar—H),7.16(m,1H,C—H),7.29(m,1H,Ar—H),7.53(m,1H,Ar—H),10.03(brs,1H,N—H)。13C NMR(100 MHz, CDCl3), δ:102.2,118.3,119.2,119.5,120.9,124.7,131.1,135.6。元素分析,C8H6ClN,实测值(计算值),%:C 63.41 (63.39);H 3.98(3.99)。
1c:3.83 g白色针状固体7-溴吲哚,收率70.94%,含量99.47%,熔点42~43 ℃(文献值[38]41~43 ℃)。1H NMR(DMSO, 400 MHz),δ:6.56(dd,J=2.8Hz,C—H),7.01(m,1H,C—H),7.31(d,J=7.2Hz,1H,Ar—H),7.37(t,J=8.5Hz,1H,Ar—H),7.57(m,1H,Ar—H),9.93(brs,1H,N—H)。13C NMR(100 MHz, CDCl3),δ:98.8,101.9,119.4,121.0,124.1,124.9,131.1,135.7。元素分析,C8H6BrN,实测值(计算值),%:C 49.00 (49.01);H 3.05(3.08)。
2 结果与讨论
2.1 反应温度和反应时间对酰胺化反应收率的影响
实验发现,按Sandmeyer异亚硝基乙酰替苯胺靛红合成法制备7-卤代靛红过程,第一步乙酰化反应温度和反应时间对产品收率有较大影响。邻卤代苯胺、盐酸羟鞍、水合氯醛、无水硫酸钠等物料配比不变,TLC监控原料至邻卤代苯胺反应完全,按上述步骤分别制备3a、3b、3c,考察乙酰化反应温度和时间对产品收率的影响,结果如表1所示。
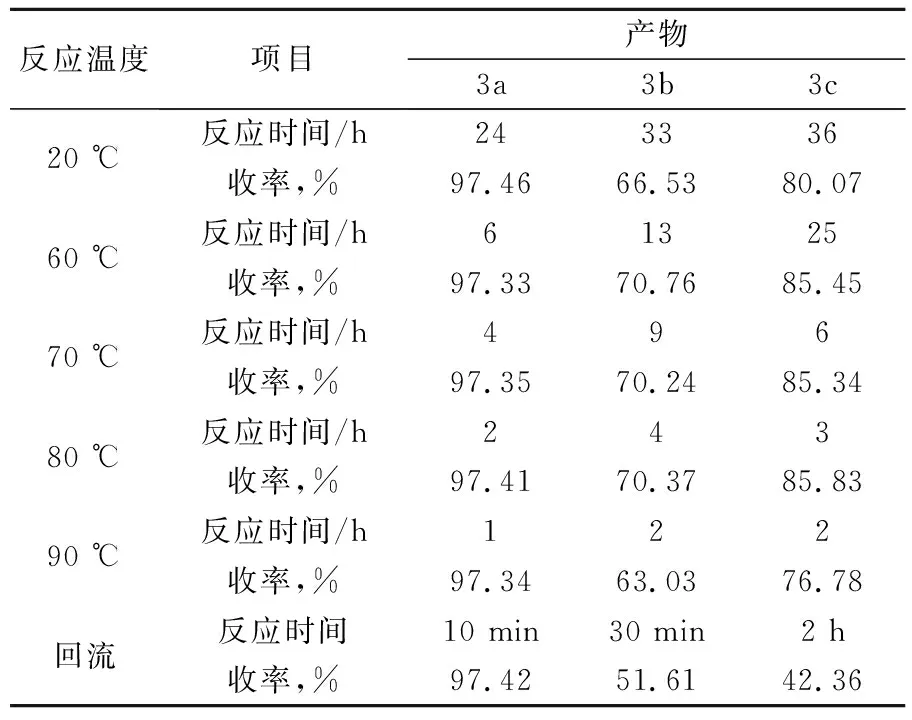
表1 反应温度、反应时间对乙酰化反应收率的影响
由表1可见,乙酰化反应过程,监控TLC至原料反应完全,随着温度升高,反应时间会缩短,但反应时间过长和温度过高,都可能导致收率下降。制备3a时,随着反应温度升高,苯胺原料反应完全时的收率基本接近,但反应时间明显缩短,回流反应时仅需10 min就有较高的收率,综合考虑选择回流反应10 min。但制备3b,3c时,20 ℃反应时间需要分别达33 h,36 h时原料才反应完全,且有灰黑色焦油状的副产物出现,导致收率较低;达到90 ℃和回流温度反应时,产生大量焦状物,收率反而明显下降,综合考虑,分别选择在80 ℃反应 4 h和3 h。
2.2 反应温度对成环反应收率的影响
乙酰苯胺闭环合成7-卤代靛红时,由于中间体在硫酸中溶解放热,加料时会放出大量热量,加料过快,会导致飞温现象,所以加料时加料速度要适中,保持温度在 60~70 ℃,并且通常1 h内加料完毕。浓硫酸具有强氧化性和强脱水性,如加料完毕后的反应温度过高,会导致中间体原料碳化而影响靛红收率,但温度过低则无法发生成环反应,因此成环反应温度对靛红收率有较大影响。其他条件不变,改变不同反应温度,考察反应温度对成环反应收率的影响,结果如图2所示。
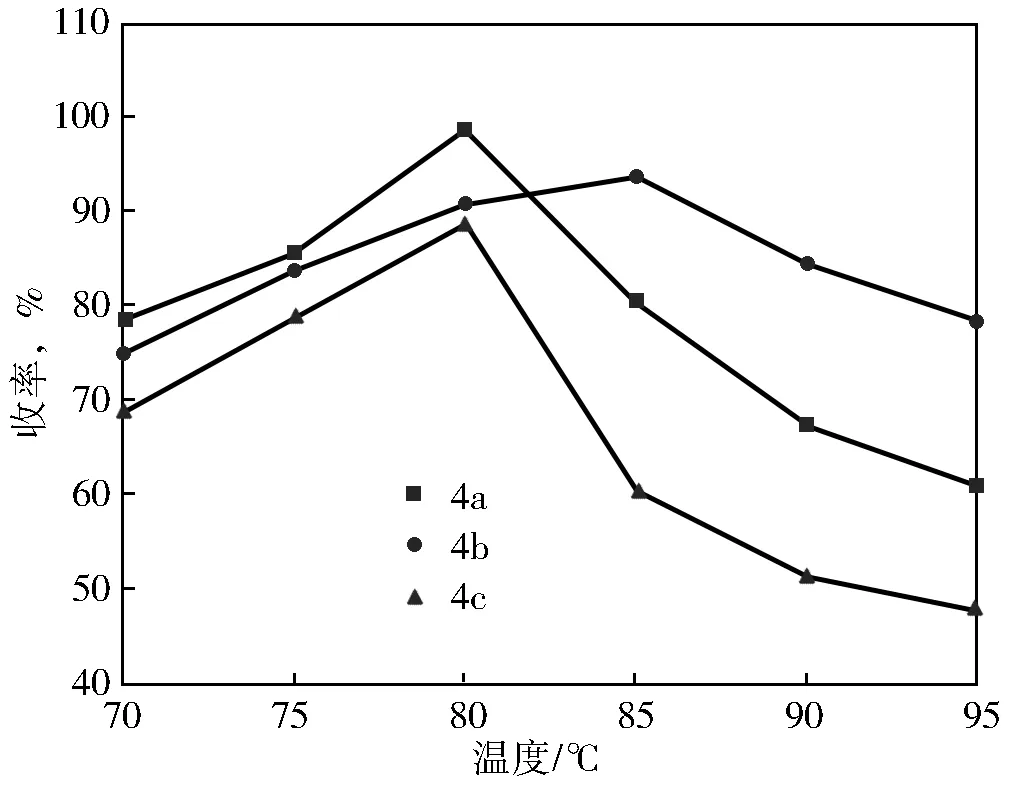
图2 反应温度对成环反应收率的影响
从图2可见,制备4a、4b和4c时,当反应温度分别为80 ℃、85 ℃和80 ℃时闭环反应收率最高,而当反应温度继续升高,收率反而下降,为此最佳成环反应温度分别为80 ℃、85 ℃和80 ℃。
2.3 还原剂种类对吲哚收率的影响
由于三氟化硼乙醚容易水解,所以由靛红还原制备吲哚步骤中必须采用无水操作,所使用的四氢呋喃必须严格除水,现蒸现用。滴加三氟化硼乙醚后,反应液会有黏稠的糊状物产生,所以需要密封体系强烈搅拌,使用磁力搅拌可能会导致搅拌失效,收率下降。还原步骤中,先有硼氢化钠与三氟化硼乙醚反应产生乙硼烷,真正起还原作用的是乙硼烷。乙硼烷也可选用市售的乙硼烷四氢呋喃溶液。反应物配比等反应条件不变,对比两种硼烷对还原反应收率的影响,结果见表2所示。
由表2可知,直接使用制备好的乙硼烷/四氢呋喃和硼氢化钠/三氟化硼乙醚还原体系,收率基本相当,虽然使用市售的制备好的乙硼烷可减少一些操作步骤,但是乙硼烷易挥发,有剧毒,易燃,对环境和人员都有较大的危险性,不适合工业化应用,所以为了安全环保,使用硼氢化钠/三氟化硼乙醚还原体系。

表2 还原剂种类对吲哚收率的影响
2.4 三氟化硼乙醚用量对还原反应收率的影响
以干燥的四氢呋喃为溶剂,n(硼氢化钠)∶n(1a~1c)=3∶1,反应10 h,考察三氟化硼乙醚用量对还原反应收率的影响,结果如图3所示。

图3 三氟化硼乙醚用量对还原反应收率的影响
由图3可知,3个还原反应的收率都随n(三氟化硼乙醚) ∶n(1a~1c)增大而增加,当n(三氟化硼乙醚)∶n(1a~1c)=2.25∶1时收率较高,再增大三氟化硼乙醚用量,收率增加不明显。综合考虑,选择n(三氟化硼乙醚)∶n(1a~1c)=2.25∶1较佳。
2.5 硼氢化钠用量对还原反应收率的影响
以干燥的四氢呋喃为溶剂,n(三氟化硼乙醚)∶n(1a~1c)=2.25∶1,反应10h,考察硼氢化钠用量对还原反应收率的影响,结果如图4所示。

图4 硼氢化钠用量对还原反应收率的影响
由图4可知,3个还原反应的收率都随n(硼氢化钠)∶n(1a~1c)增大而增加,当n(三氟化硼乙醚)∶n(1a~1c)=3.0∶1时收率较高,再增大硼氢化钠用量,收率变化不大。另外由于硼氢化钠遇水易燃,过多地使用硼氢化钠会给后处理增加麻烦。综合考虑,选择n(硼氢化钠)∶n(1a~1c)=3.0∶1为宜。
3 结 论
以邻卤代苯胺和水合氯醛、盐酸羟胺为原料,通过Sandmeyer异亚硝基乙酰替苯胺合成法制备得7-卤代靛红,再经硼氢化钠/三氟化硼乙醚体系还原制备得7-卤代吲哚。实验考察了酰胺化反应温度和反应时间、成环反应温度、还原体系及还原剂用量对反应收率的影响,结果表明,制备7-氟吲哚1a, 7-氯吲哚1b,7-溴吲哚1c时,较佳的酰胺化反应温度和反应时间分别为回流10 min、80 ℃ 4 h和80 ℃ 3 h,较佳的成环反应温度分别为80、85和80 ℃,较优的还原体系为硼氢化钠/三氟化硼乙醚体系,较优的还原剂用量为n(硼氢化钠)∶n(三氟化硼乙醚)∶n(卤代靛红)=3.0∶2.25∶1。由7-卤代靛红还原制备7-卤代吲哚的工艺,与现有7-卤代吲哚合成工艺相比,具有原料成本低,反应条件温和,安全高效,操作简便等优点,适合批量制备7-卤代吲哚。
参 考 文 献
[1] Yao C,Song J,Chen C T,et al.Synthesis and biological evaluation of novel C-indolylxylosides as sodium-dependent glucose co-transporter 2 inhibitors[J].Eur J Med Chem,2012(55):32-38.
[2] Choi-Sledeski Y,Liang G,Poli G.Preparation of spiropiperidine benzylamines as therapeutic beta-tryptase inhibitors:WO,2011079103 A1[P].2011-06-30.
[3] Minatti Ana,Low J,Allen J,et al.Preparation of perfluorinated 5,6-dihydro-4H-1,3-oxazin-2-amine compounds as β-secretase inhibitors and methods of use: WO,2014134341 A1[P].2014-09-04.
[4] Sato K,Sugimoto H,Rikimaru K,et al.Discovery of a novel series of indoline carbamate and indolinylpyrimidine derivatives as potent GPR119 agonists[J].Bioorg Med Chem,2014,22(5):1649-1666.
[5] Seltzman H H,Shiner C,Hirt E E,et al.Peripherally selective cannabinoid 1 receptor (CB1R) agonists for the treatment of neuropathic pain[J].J Med Chem,2016,59(16),7525-7543.
[6] Spigelman I,Seltzman H H,Shiner C.Preparation of substituted 1H-indene and 1H-indole compounds as peripherally-acting cannabinoid receptor agonists for chronic pain: WO,2014015298 A1[P].2014-01-23.
[7] Narayanan R,Miller D D,Ponnusamy T,et al.Preparation of novel indole,indazole,benzimidazole,indoline,quinolone,isoquinoline,and carbazole selective androgen receptor degrader (SARD) ligands: WO,2016172358 A1[P].2016-10-27
[8] Fox B M,Beck H P,Roveto P M,et al.A Selective prostaglandin E2 receptor subtype 2 (EP2) antagonist increases the macrophage-mediated clearance of amyloid-beta plaques[J].J Med Chem,2015,58(13): 5256-5273.
[9] Maloney D J,Jadhav A,Bantukallu G R,et al.Pyrazole derivatives as inhibitors of lactate dehydrogenase and their preparation: WO,2016109559 A2[P].2016-07-07.
[10] Tatlock J H,McAlpine I J,Tran-Dube M B,et al.Preparation of substituted nucleoside derivatives useful as anticancer agents: US,20160244475 A1[P].2016-08-25.
[11] Yu P,Wang Y Q,Teng Y O.Tetrahydropyrano[3,4-b]indole derivatives as antitumor agents and their preparation,pharmaceutical compositions and use in the treatment of diseases: CN,103965207 A[P].2014-08-06.
[12] Romero F A,Magnuson S,Pastor R,et al.4,5,6,7-Tetrahydro-1H-pyrazolo[4,3-c]pyridin-3-amine compounds as CBP and/or EP300 inhibitors and their preparation: WO,2016086200 A1[P].2016-06-02.
[13] Mistry B,Keum Y S,Kim D H.Synthesis,antioxidant and anticancer screenings of berberine-indole conjugates[J].Res Chem Intermed,2016,42(4): 3241-3256
[14] Denis J,Jolivalt C,Maurin M,et al.Preparation of bis-indole derivatives useful as antibacterials: WO,2013014104 A1[P].2013-01-31.
[15] Renner J,Dietz J,Grote T,et al.Preparation of imidazoles and triazoles as agrochemical fungicides: WO,2010028974 A1[P].Mar 18,2010-03-18.
[16] David N,Pasceri R,Kitson R,et al.Formal total synthesis of diazonamide A by indole oxidative rearrangement[J].Chem A Eur J,2016,22(31):10867-1087.
[17] Adams S E,Parr C,Miller D,et al.Potent inhibition of Ca2+-dependent activation of calpain-1 by novel mercaptoacrylates[J].Med Chem Comm,2012,3(5):566-570.
[18] Chen J C,Zhang Z K,Liu S J,et al.One-pot tandem synthesis of 2,3-unsubstituted indoles,an improved Leimgruber-Batcho indole synthesis[J].RSC Adv,2014,4(9):4672-4675.
[19] Batcho A D,Leimgruber W.Indoles from 2-methylnitrobenzenes by condensation with formamide acetals followed by reduction: 4-Benzyloxyindole[J].Org Synth Coll,1990,7:34.
[20] Xia C H,Zhang Z K,Liu S J.“One-pot” method for synthesizing substituted indole-like compound: CN,102924359A[P].2013-02-13.
[21] Robbins D W,Boebel T A,Hartwig J F.Iridium-Catalyzed,Silyl-Directed Borylation of Nitrogen-Containing Heterocycles[J].J Am Chem Soc,2010,132(12): 4068-4069.
[22] Yamada Y,Arima S,Okada C,et al.Preparation of 7-halo-indoles by thallation ofN-formylindoline and their attempted use for synthesis of the right-hand segment of chloropeptin[J].Chem Pharm Bull,2006,54(6):788-794.
[23] Joule J A.Product class 13: indole and its derivatives[J].Science of Synthesis,2001,10:361-652.
[24] Chen Z G,Cohen M P,Fisher M J,et al.Preparation of N-(2-arylethyl) benzylamines as antagonists of the 5-HT6 receptor: WO,2002078693 A2[P]: 2002-10-10.
[25] Farina V,Krishnamurthy V,Scott W.The Stille reaction[M].Hoboken: Division of Chemical Education,1997: 50.
[26] Thansandote P,Hulcoop D G,Langer M,et al.Palladium-catalyzed annulation of haloanilines and halobenzamides using uorbornadiene as an acetylene synthon: a route to functionalized indolines,isoquinolinones,and indoles[J].J Org Chem,2009,74(4):1673-1678.
[27] Schlosser M,Ladenberger V.Basen-induzierte eliminierungen,III.der einflu von stereochemie und substituenten aufdie reaktivität von vinylchloriden[J].Chem Ber,1967,100:3901-3915.
[28] Schlosser M,Ginanneschi A,Leroux F.In search of simplicity and flexibility: a rational access to twelve fluoroindolecarboxylic acids other sources[J].Eur J Org Chem,2006,13:2956-2969.
[29] Bratulescu G.A new and efficient one-pot synthesis of indoles[J].Tetrahedron Lett,2008,49(6):984-986.
[30] Uchikawa O,Mitsui K,Asakawa A,et al.Condensed pyrazole derivatives,process for producing the same and use thereof: US,6949648B2[P].2005-09-27.
[31] Holt S J,Sadler P W.Studies in Enzyme Cytochemistry.II.Synthesis of indigogenic substrates for esterases[J]. Proc Roy Soc Lond B Bio,1958,148:481-488.
[32] Allen F L,Brunton J C,Suschitzky H. Heterocyclic fluorine compounds.Part II.bz-monofluoroindoles[J]. J Chem Soc,1955:1283-1284.
[33] Baruffini.Synthesis of pyrazole derivatives[J].Farmaco,Edizione Scientifica,1967,22: 590-598.
[34] Matheus M, Violante F, Garden S, et al. Isatins inhibit cyclooxygenase-2 and inducible nitric oxide synthase in a mouse macrophage cell line[J].Eur J Pharmacol,2007,556(13):200-206.
[35] Bratulescu G.A new and efficient one-pot synthesis of indoles[J].Tetrahedron Lett,2008,49(6):984-986.
[36] Beauchard A,Laborie H,Rouillard H,et al.Synthesis and kinase inhibitory activity of novel substituted indigoids[J].Bioorg Med Che,2009,17(17):6257-6263.
[37] Matesic L,Locke J M,Vine K L,et al.Synthesis and anti-leukaemic activity of pyrrolo[3,2,1-hi]indole-1,2-diones,pyrrolo[3,2,1-ij]quinoline-1,2-diones and other polycyclic isatin derivatives[J].Tetrahedron,2012,68(34):6810-6819.
[38] Fan-Chiang T,Wang H,Hsieh J.Synthesis of phenanthridine skeletal Amaryllidaceae alkaloids [J].Tetrahedron,2016,72(36):5640-5645.
猜你喜欢
云南化工(2021年11期)2022-01-12 06:06:10
昆明医科大学学报(2021年2期)2021-03-29 07:42:14
商情(2020年47期)2020-12-15 06:53:14
科技视界(2019年12期)2019-06-20 01:34:27
百家讲坛(2019年24期)2019-04-24 00:27:35
农业与技术(2018年20期)2018-11-16 12:31:54
环境与发展(2018年3期)2018-05-10 11:21:48
湖南理工学院学报(自然科学版)(2017年4期)2018-01-25 06:01:08
无机盐工业(2017年5期)2017-05-25 00:37:34
无机化学学报(2014年7期)2014-02-28 17:32:16
