
超支化聚氨酯多元醇的反应动力学
2018-06-05 05:38:50罗玉梅叶仲斌白小东
石油化工 2018年5期
罗玉梅,叶仲斌,白小东,周 松
(1.西南石油大学 油气藏地质及开发工程国家重点实验室,四川 成都 610500;2.西南石油大学 材料科学与工程学院,四川 成都 610500)
超支化聚氨酯具有高官能度分子结构,与相应的线型分子相比,超支化聚氨酯具有较低的熔融和溶液黏度。超支化聚氨酯的合成方法主要有单-单体合成法,包括光气法[1]、叠氮化合物法[2]、高选择性化学反应法[3]、Curtius反应法[4]等;双单体合成法[5-8],包括A2+B3法,A2+bB2法等;超支化聚合物扩链法[9-11]。DSC 法[12-13]、FTIR 法[14]、拉曼光谱法[15]常常被用于聚氨酯的反应动力学研究,然而,在聚氨酯反应动力学的研究中,试样峰在较宽范围内的重叠会影响动力学分析,FTIR表征中试样的厚度对定量分析也有较大影响,而且每时刻反应物仅有微量的变化,以至于上述方法不能准确地检测和描述聚氨酯的反应动力学。
本工作以异佛尔酮二异氰酸酯(IPDI)和三羟甲基丙烷(TMP)为单体,采用溶液聚合法,在无催化剂的条件下合成了端羟基超支化聚氨酯多元醇(HBPU),并采用二正丁胺滴定法对反应进行了动力学研究。
1 实验部分
1.1 试剂
IPDI:分析纯,Sigma-Aldrich贸易有限公司;TMP:分析纯,阿拉丁试剂有限公司;N,N-二甲基甲酰胺(DMF)、乙醇、丙酮、四氢呋喃、二正丁胺、吡啶:分析纯,成都市科龙化工试剂厂。
1.2 实验步骤
将三颈瓶置于油浴中,装上冷凝管、搅拌器、温度计,加入0.05 mol TMP和30 mL DMF搅拌均匀,然后加入0.15 mol IPDI,反应60 min后得中间产物T3I,再加入20 mL DMF和0.15 mol TMP,反应120 min,冷却至40 ℃,将产物放入茄形烧瓶中,用旋转蒸发仪在115 ℃、40 r/min的转速下蒸馏5 h,得到无色透明的产物。
1.3 表征
采用美国Nicolet公司Thermal Nicolet 6700型傅里叶变换红外光谱仪进行FTIR表征,涂膜法,测试范围650~4 000 cm-1,分辨率4 cm-1,扫描速率48 cm/s;采用美国Waters科技有限公司Waters BreezeTM2型高效液相色谱仪(凝胶渗透色谱柱)对相对分子质量及其分布进行测定,流动介质为四氢呋喃,测试温度30 ℃,标样为聚苯乙烯。
二正丁胺滴定法:取1~1.5 g试样于250 mL锥形瓶中,加入20 mL丙酮使试样溶解,再用移液管移取10 mL二正丁胺-丙酮溶液,使瓶内液体混合均匀,静置20 min,加入30 mL乙醇和4滴溴甲酚绿指示剂。用0.5 mol/L的HCl标准溶液滴定,溶液由蓝色变成黄色15 s不变为终点,同时做空白实验。异氰酸酯(NCO)与过量二正丁胺反应生成取代脲,用HCl标准溶液滴定剩余的二正丁胺,再与空白实验所消耗的HCl结果相比,即可计算出NCO基团含量。
2 结果与讨论
2.1 聚合反应动力学方程的建立
2.1.1 预聚反应阶段
生成的氨基甲酸酯(NHCOO)基团含量较低,NCO基团含量较高,反应过程见式(1)。
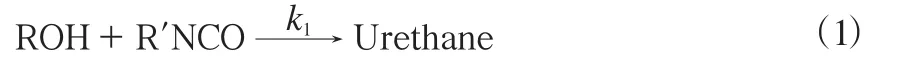
式中,k1为预聚反应的反应速率常数,kg/(mol·min)。
反应速率可用式(2)表示。

式中,t为预聚反应时间,min;c为预聚反应t时刻体系中i基团的质量摩尔浓度,mol/kg。
当反应平衡时,有crNCO=crOH成立,设crNCO=c0NCO-cNCO,crOH=c0OH-cOH,结合反应条件可得式(3)。
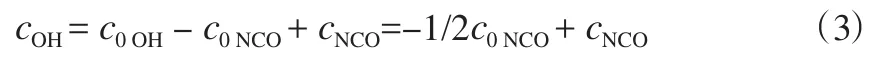
式中,c0为预聚反应体系中i基团的初始质量摩尔浓度,mol/kg;cr为预聚反应体系中已反应的i基团的质量摩尔浓度,mol/kg。
将式(3)代入式(2),得式(4)。
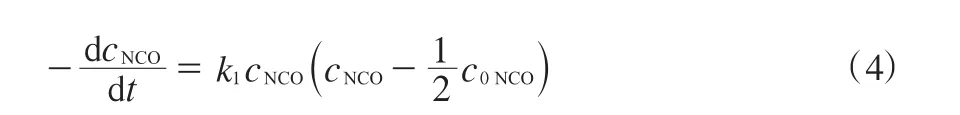
设预聚阶段NCO的转化率为α,则

对时间t求导并整理可得式(6)。
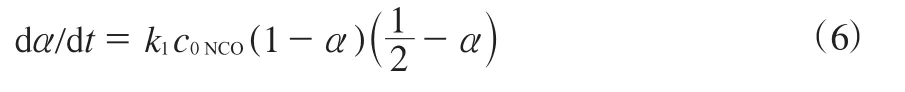
积分可得式(7)。
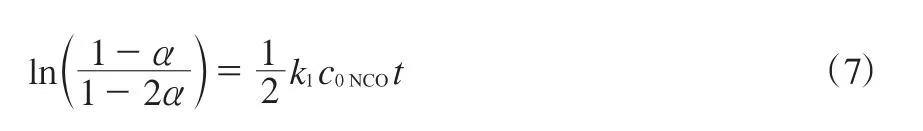
2.1.2 聚合反应阶段
NHCOO基团的含量较高,NCO基团含量较低,聚合反应伴随着NHCOO的自催化效应,反应过程见式(8)。
式中,k2为聚合反应的反应速率常数,kg2/(mol2·min)。
整理后,反应速率见式(9)。

式中,t′为聚合反应时间,min;c′为聚合反应t′时刻体系中i基团的质量摩尔浓度,mol/kg;c0′为聚合反应体系中i基团的初始质量摩尔浓度,mol/kg;cr′为聚合反应体系中已反应的i基团的质量摩尔浓度,mol/kg。
设聚合阶段NCO的转化率为α′,则

整理可得式(11)
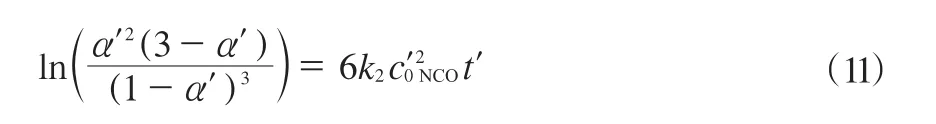
由式(7)和式(11)可知,等式的左边项和右边项中的反应时间呈线性关系,由直线斜率可得到反应速率常数,再根据Arrhenius方程可求出反应的活化能。
2.2 预聚反应动力学的数据处理
图1为预聚反应中不同反应温度下NCO转化率随时间的变化。由图1可知,反应温度越高,转化率增大越快。80 ℃时,转化率在5 min内已达到30%,反应1 h后已接近反应完全的转化率理论值(50%)。
预聚反应中不同反应温度下ln[(1-α)/(1-2α)]与t的关系见图2。由图2可知,两者存在较好的线性关系,与实验结果基本相符,说明反应动力学模型比较合理。
根据直线斜率可得到不同温度下的预聚反应速率常数和活化能,见表1。
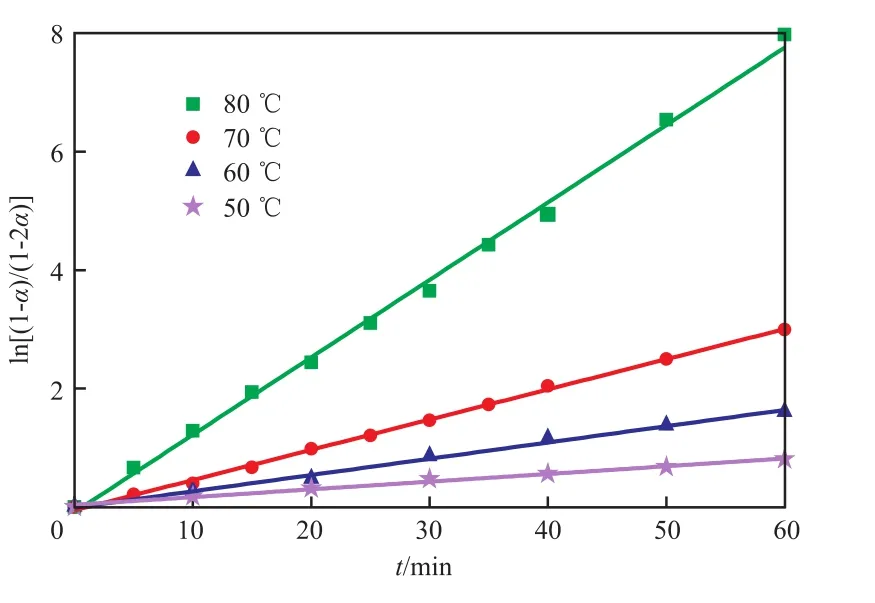
图2 预聚反应中不同温度下ln[(1-α)/(1-2α)]~t的关系Fig.2 The relationship of ln[(1-α)/(1-2α)]and t at different temperatures in the pre-polymerization reaction.
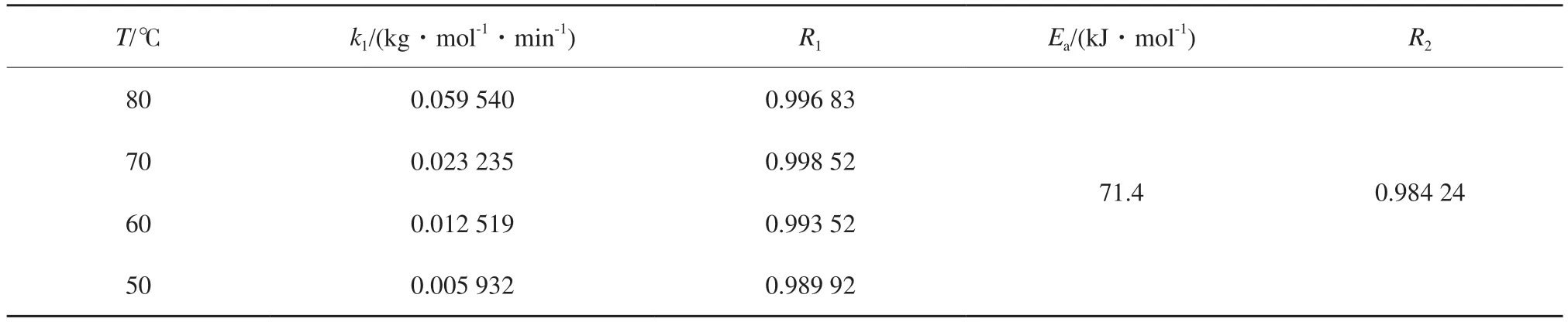
表1 不同温度下的预聚反应速率常数和活化能Table 1 Pre-polymerization rate constants(k1) and activation energy at different temperatures
随着反应温度的升高,预聚反应速率增大,80 ℃时反应速率常数比50 ℃时增加了近10倍。可见,预聚反应速率的大小主要取决于反应温度。
根据Arrhenius方程,对lnk1和1/T做图,如图3所示。TMP和IPDI反应生成中间产物T3I的反应活化能为71.4 kJ/mol。
2.3 中间产物T3I的FTIR谱图
图4为中间产物T3I的FTIR谱图。由图4可知,3 444 cm-1处是O—H伸缩振动吸收峰,峰较宽是由于OH和NH形成了氢键;50,60,70 ℃下反应1 h后O—H伸缩振动吸收峰仍然存在;但80 ℃下反应1 h后该峰已经消失,说明此时体系中的羟基已反应完全。
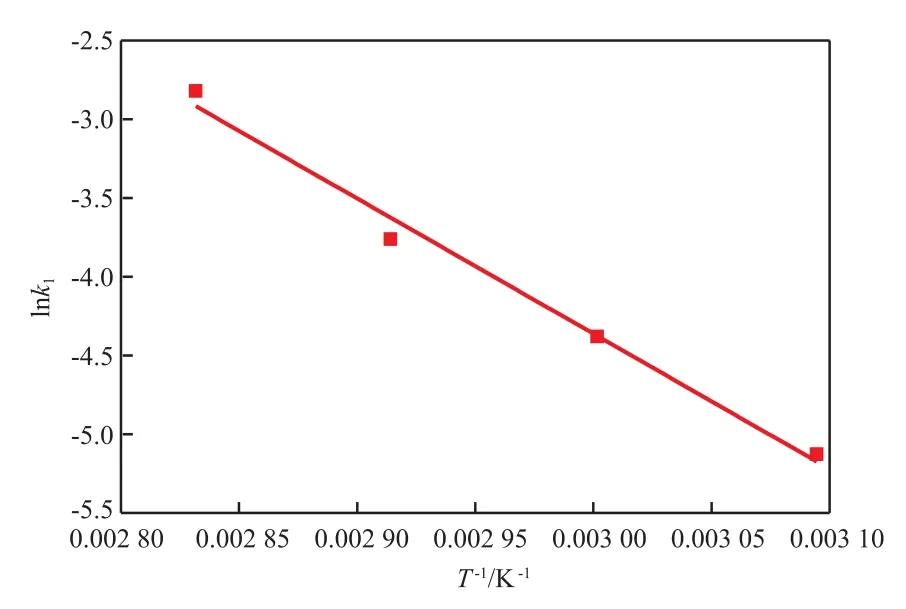
图3 预聚反应速率常数k1随温度的变化Fig.3 Arrhenius plots of pre-polymerization reaction rate constant versus temperature.
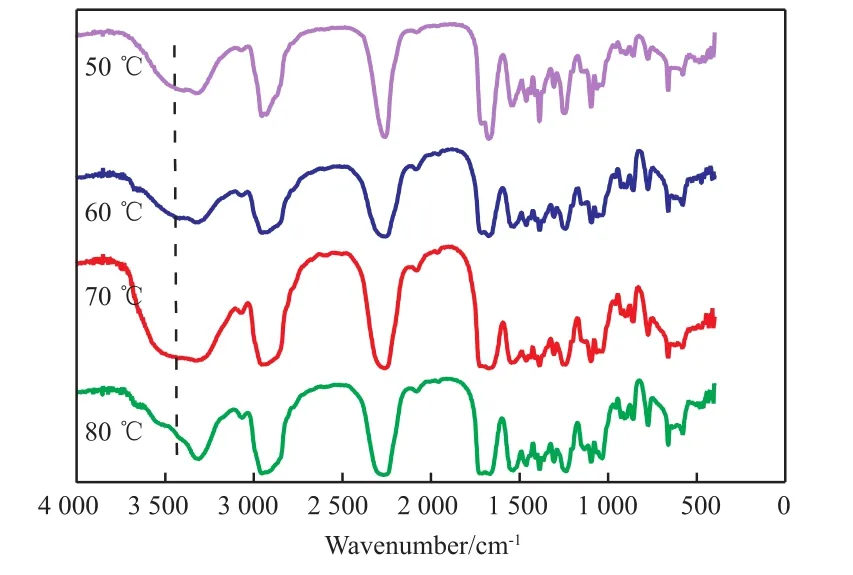
图4 中间产物T3I的FTIR谱图Fig.4 FTIR spectra of intermediate product T3I.Reaction conditions referred to Fig.1.
2.4 聚合反应动力学的数据处理
图5为聚合反应中不同温度下NCO的转化率随时间的变化。由图5可知,80 ℃下反应约30 min后,NCO转化率已达80%,反应2 h后NCO转化率接近100%。各温度下HBPU聚合反应均未出现凝胶。
聚合反应中不同反应温度下ln[α′2(3-α′)/(1-α′)3]与t′的关系见图6。由图6可知,两者呈现较好的线性关系,说明反应动力学模型比较合理。
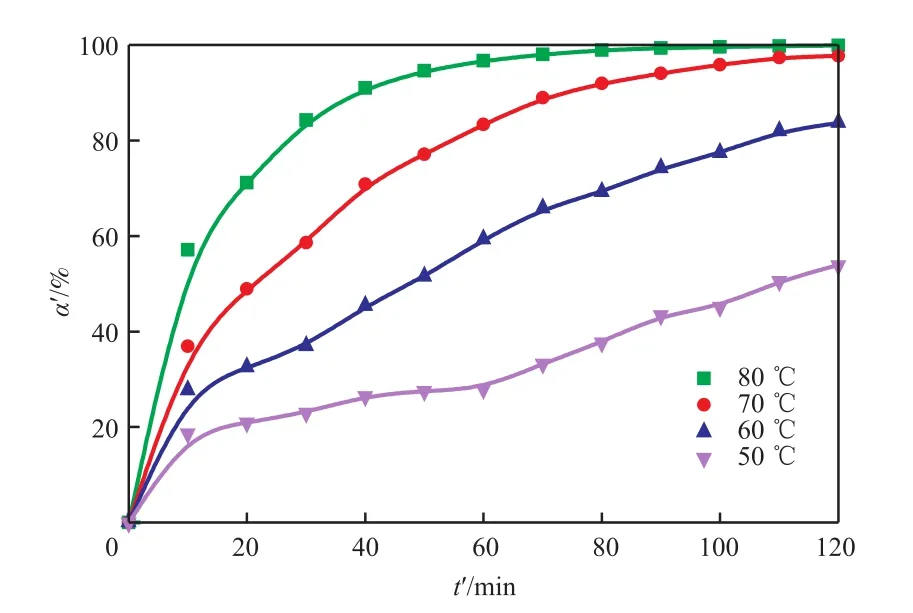
图5 聚合反应中不同温度下NCO的转化率随时间的变化Fig.5 Plots of the NCO conversion versus time at different temperatures in the polymerization reaction.
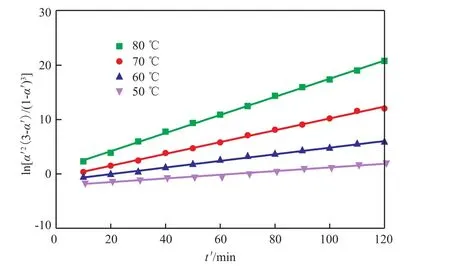
图6 聚合反应中不同温度下ln[α′2(3-α′)/(1-α′) 3]~t′的关系Fig.6 The relationship of ln[α′2(3-α′)/(1-α′) 3] and t′ at different temperatures in the polymerization reaction.
根据直线斜率可得到不同温度下的聚合反应速率常数和活化能,见表2。由表2可知,与预聚反应相似,聚合反应速率随反应温度的升高而增大,80 ℃时反应速率常数比50 ℃时的增加近5倍。与预聚反应相比,反应温度对聚合反应速率常数的影响较小,活化能数值也说明了这一点。
根据Arrhenius方程,对lnk2和1/T做图,如图7所示。结合表2和图7可知,中间产物T3I与TMP反应的活化能E′a为51.1 kJ/mol。聚合反应的活化能比预聚反应的活化能小,说明预聚反应受温度的影响更大。可见,在没有催化剂的情况下,NHCOO的自催化效应在HBPU聚合反应中起着非常重要的作用。
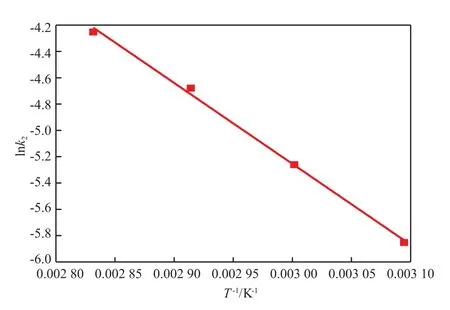
图7 聚合反应速率常数k2随温度的变化Fig.7 Arrhenius plots of polymerization reaction rate constant versus temperature.
2.5 聚合产物HBPU的FTIR谱图
图8为HBPU的FTIR谱图。
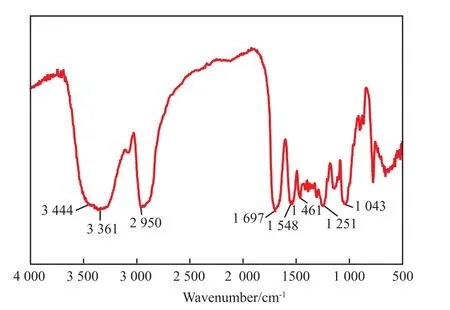
图8 HBPU的FTIR谱图Fig.8 FTIR spectrum of the hyperbranched polyurethane polyol.
由图8可知,3 444 cm-1处的强吸收峰归属于羟基的O—H伸缩振动,3 361 cm-1处的吸收峰是由N—H键的伸缩振动引起的,此处的峰较宽是O—H和N—H吸收峰重叠造成的。2 950 cm-1处的吸收峰是由CH,CH2,CH3的C—H伸缩振动产生的。1 697 cm-1处的强峰是羰基的伸缩振动吸收峰。1 548 cm-1处的吸收峰是由N—H键的弯曲振动引起的。1 461 cm-1处的吸收峰是C—H弯曲振动和C—N伸缩振动产生的。1 251 cm-1处的吸收峰是O==C—O结构中C—O键的伸缩振动吸收峰。1 043 cm-1处的吸收峰归因于C==OH结构中的C—O键伸缩振动。2 200 cm-1处没有明显的吸收峰出现,说明NCO基团已经反应完全。综上所述,产物中含有NHCOO基团和羟基,不含有NCO基团。所测得的HBPU羟值(KOH)为399 mg/g,数均相对分子质量为1 348,重均相对分子质量1 702,多分散系数1.26,可推断产物的分子结构并非呈规则的树枝状,而是低代数的HBPU。
2.6 HBPU的反应动力学分析
氨酯化反应通常只在有限范围内遵循动力学方程,但在HBPU的反应动力学中可在较大范围内符合动力学方程。这是因为超支化聚氨酯本身具有很强的极性,聚合过程中体系的极性变化很小,而在线型聚氨酯的聚合过程中,早期体系主要由NCO类和醇类反应物构成,极性较大;随着反应的进行,体系逐渐由极性较弱的线型NHCOO构成,这种体系极性的变化对反应速率常数有较大的影响。另外,超支化聚氨酯具有较低的熔体和溶液黏度,反应过程中体系的黏度变化不大,即使反应后期体系黏度也不高,对反应的影响较小。在预聚反应阶段NCO转化率较低时,体系中生成的NHCOO基团较少,体系中仅存在NCO和OH的反应,反应动力学可归为二级反应。在聚合反应阶段NCO转化率较高,体系中生成的NHCOO基团较多时,NHCOO基团影响不可忽视;由于OH和生成的NHCOO之间形成氢键,促进了OH和NCO的反应,具有自催化效应,此时反应动力学遵循三级反应。
3 结论
1)HBPU的反应分为预聚反应和聚合反应两步进行。预聚反应生成中间产物T3I的最佳反应条件为n(NCO)∶n(OH)= 2∶1,反应温度80 ℃,反应时间60 min。聚合反应的最佳条件为n(NCO)∶n(OH)= 1∶3,反应温度 80 ℃,反应时间120 min。
2)产物HBPU中含有NHCOO基团和羟基,不含有NCO基团。所测得的HBPU羟值(KOH)为399 mg/g,数均相对分子质量为1 348,重均相对分子质量1 702,多分散系数1.26,可推断产物的分子结构是低代数的HBPU。
3)HBPU的预聚反应动力学遵循二级反应,反应活化能为71.4 kJ/mol,聚合反应动力学遵循三级反应,反应活化能为51.1 kJ/mol。聚合反应的活化能比预聚反应的活化能小,说明预聚反应受温度的影响更大,在没有催化剂的情况下,NHCOO的自催化效应在HBPU聚合反应中起着重要的作用。
[1] Ralph S,Jean M J F. Synthesis and characterization of hyperbranched polyurethanes prepared from blocked isocyanate monomers by step-growth polymerization[J].Macromolecules,1993,26(18):4809-4813.
[2] Chickiyan S,Nasar A S. Hydroxyl- and amine-terminated hyperbranched polyurethanes using AB2-type azide monomers:Synthesis,characterization,fluorescence,and chargetransfer complexation studies[J].J Polym Sci,Part A:Polym Chem,2009,47(13):3337-3351.
[3] Rannard S P,Davis N J,Herbert I. Synthesis of water soluble hyperbranched polyurethanes using selective activation of AB2monomers[J].Macromolecules,2004,37(25):9418-9430.
[4] Taylor R T,Puapaiboon U. Polyurethane dendrimers via curtius reaction[J].Tetrahedron Lett,1998,39(44):8005-8008.
[5] Che Pengchao,He Yaning,Wang Xiaogong. Synthesis and characterization of a hyperbraned azo polymerization[J].Acta Polym Sinica,2007 (1):21-25.
[6] 陈正宇,程秀美,吴智超,等. 超支化聚合物的合成、表征及其对水性聚氨酯膜性能的影响[J].材料导报,2013,27(11):85-87.
[7] 鲁慧,刘胜波,张良均,等. 超支化水性聚氨酯羟基组分的制备及成膜性能研究[J].试验研究与应用,2012,15(3):4-7.
[8] 梅锦岗,杨建军,吴明元,等. 超支化水性聚氨酯的合成与表征[J].高分子材料科学与工程,2012,28(6):17-20.
[9] Czech P,Okrasa L,Boiteux G,et al. Polyurethane networks based on hyperbranched polyesters:Synthesis and molecular relaxations[J].J Non-Cryst Solids,2005,351(33/36):2735-2741.
[10] 刘棚滔,杨建军,吴庆云,等. 羟丙基硅油改性超支化水性聚氨酯的合成与性能[J].化工新型材料,2016,44(2):67-69.
[11] Nanda A K,Wicks D A. The Influence of the ionic concentration,concentration of the polymer degree of neutralization and chain extension on aqueous polyurethane dispersions prepared by the acetone process[J].Polymer,2006,47(6):1805-1811.
[12] Sultan W,Busnel J P. Kinetic study of polyurethanes formation by using differential scanning calorimetry[J].J Therm Anal Calorim,2006,83(2):355-359.
[13] Bina C K,Kannan K G,Ninan K N. DSC study on the effect of isocyanates and catalysts on the HTPB cure reaction[J].J Therm Anal Calorim,2004,78:753-760.
[14] Xue Shuchang,Zhang Zhiping,Ying Shengkang. Reaction kinetics of polyurethane/polystyrene interpenetrating polymer networks by infra-red spectroscopy[J].Polymer,1989,30:1269-1274.
[15] Parnell S,Min K,Cakmak M. Kinetic studies of polyurethane polymerization with Raman spectroscopy[J].Polymer,2003,44(18):5137-5144.
猜你喜欢
数学年刊A辑(中文版)(2021年1期)2021-06-09 09:32:06
皮革制作与环保科技(2020年13期)2020-03-17 07:12:20
上海建材(2019年4期)2019-05-21 03:13:04
铜仁学院学报(2018年6期)2018-07-05 09:47:34
山西大同大学学报(自然科学版)(2016年4期)2016-11-27 02:20:55
衡阳师范学院学报(2016年3期)2016-07-10 07:16:27
新高考·高一物理(2016年3期)2016-05-18 16:16:56
中国塑料(2015年8期)2015-10-14 01:10:46
橡胶工业(2015年2期)2015-07-29 08:29:40
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01 02:53:54
