
万寿菊-烟草套作体系中不同生长期烟草根际土壤微生物多样性研究
2018-06-05 05:56:49夏体渊陈泽斌刘佳妮王定康徐胜光
西南农业学报 2018年4期
夏体渊,陈泽斌,苏 源,刘佳妮,余 磊,王定康,徐胜光
(1.昆明学院农学院,云南 昆明 650214;2.云南省高校特色生物资源开发与利用重点实验室,云南 昆明 650214;3.云南省都市特色农业工程技术研究中心,云南 昆明 650214;4.云南省高校生物炭工程研究中心,云南 昆明 650214)
【研究意义】烟草是中国重要的经济作物,它在国民经济中占有重要的地位。中国人口越来越多,田地越来越少,烟草轮作的条件越来越受到限制[1],烟草每年连作引起的经济损失每年高达40亿元[2]。连作影响根际土壤有益生物的生存,进而影响有机营养物质的转化和传递,同时增加了一些致病微生物的富集,一系列连锁反应严重影响了烟叶的品质和产量,加重了烟草作物的各种病害的发生。所以探明连作障碍的原因并采取有效措施减缓连作障碍已成为目前的研究热点。作物根际土壤微生物是土壤生态系统中最活跃的组成部分,它是土壤生态系统中能量流、物质循环和信息传递的主要参与者,因此从根际微生态系统的角度研究障碍具有重要的理论和实践意义[3]。【前人研究进展】目前关于连作对作物根际土壤微生物影响的研究已有报道,黄瓜连作后,有益微生物受到限制,有害生物得以迅速繁殖,导致病害加重[4]。岳冰冰等利用BiologTM微平板检测技术研究表明,烤烟连作明显降低了土壤微生物碳源的利用率,削弱了微生物群落的代谢活性[5]。目前已有研究表明通过不同作物间的轮套作来改善植物根际微环境,在一定程度上可以减轻作物病害。冯俊喜等研究表明烟草—小麦套作体系下微生物群落指数增大[6],烟草—小麦套作能减低烤烟马铃薯Y病毒的发病率,与甘薯套种能有效减轻烟草病毒病的危害[7]。烟草与大豆套作能提高烟田根际微生物的多样性,增加有益菌群的数量,减轻土传病害的发生程度[8]。烟蒜套作较单作烤烟根际土壤速效养分供应能力较强,特别是对磷活化利用,能提高烟叶产量[9-10]人们已通过研究利用合理的套作体系对控制连作障碍也有了一定成效,贾兰虹等[11]研究表明通过万寿菊与圆葱套作能使万寿菊和圆葱的病菌侵染都得到遏制,有效提高各自产值;魏环宇等[12]通过万寿菊与当归的套作研究表明万寿菊可显著影响根际土壤中真菌的种类及群落结构组成。孙建波[13]等采用Real-time PCR和T-RFLP技术对巴西蕉不同生长期根际细菌数量和多样性的研究表明,随着生长期的增加,细菌数量呈现先增加后减少,而细菌多样性呈逐渐减少趋势。【研究切入点】但迄今为止关于万寿菊—烟草套作体系中烟草不同生长期根际土壤微生物种类变化的研究尚未见报道。【拟解决的关键问题】本研究采用近年来兴起的Illumina MiSeq第二代测序技术全面而准确地对万寿菊—烟草套作体系下不同生长时期根际土壤微生物种类组成进行研究,相比变性凝胶梯度电泳和构建克隆文库技术,无需构建克隆;且通量高,可对混合物直接进行测序,节约了98 %以上的时间。本研究旨在探明万寿菊—烟草套作体系下根际土壤微生物群落变化情况,从根际微生态系统的角度探明烟草—万寿菊套作体系的防病机理,为今后万寿菊与其他作物的套作应用提供理论依据。

表1 样品编号
1 材料与方法
1.1 试验材料
供试烟草为云烟87,2016年4月22-24日移栽于宣威市落水镇试验田,每棵烟苗旁播种3颗万寿菊种子,以单作烟草为对照。采用5点取样法于2016年5月中旬至9月分别采集团棵期、旺长期、现蕾期、成熟期4个时期烟草单作、万寿菊-烟草套作体系烟草根际土壤,样品编号见表1,采集后立即放入保鲜袋中,24 h内进行土壤DNA提取。
1.2 总DNA提取
参照Omega公司的D5625-01 Soil DNA Kit土壤基因组DNA提取试剂盒的步骤提取各根际土壤样品DNA,用0.8 %的琼脂糖凝胶电泳检查后,用无菌水稀释至1 ng/μl。
1.3 ITS2区及16S rDNA-V4区的PCR扩增
使用带标签序列(Barcode)的ITS2区及16S rDNA-V4区特异引物3F/4R及515F/806R,使用KOD-Plus-Neo高保真PCR酶进行PCR,用2.0 %的琼脂糖凝胶电泳检测后,送北京诺禾致源生物信息科技有限公司,进行MiSeq测序。
1.4 下机数据的质控及分析
剔除标签序列(Barcode)和引物序列,Flash软件[14]进行序列拼接,并通过qiime软件[15]过滤得到的序列与Golddatabase数据库中的已知序列比对,在用UCHIME软件[16]去除嵌合体序列,得到有效序列。用Uparse软件[17]在97 %的相似性水平上划分操作分类单元,代表序列的注释采用RDP分类软件[18]和GreenGene数据库[19]完成,利用Mothur软件作稀释度曲线,计算文库覆盖率(Coverage),Chao1指数及Shannon指数计算方法参见文献[14]。使用R语言画样品OTU分析维恩图,通过Canoco软件作PCA主成分分析显示样品间的差异,利用Fastunifrac软件分析得到样品间距离矩阵,绘制基于Weighted Unifrac距离的UPGMA聚类树。
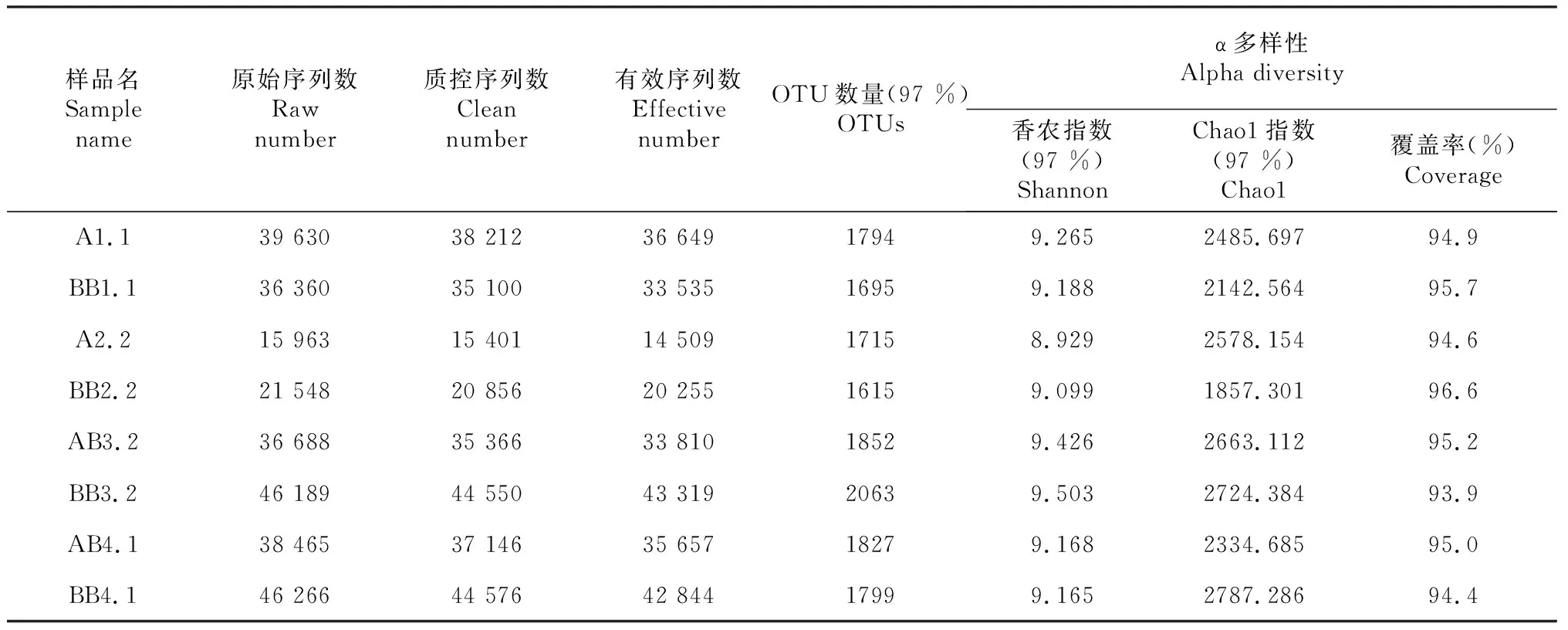
表2 样品中细菌OTU丰度和α多样性指数
2 结果与分析
2.1 测序结果与有效性验证
8个样品的细菌16S rRNA基因V4区序列被测序,得到281 109条原始序列,部分低质量序列被过滤,最终得到有效序列为260 578条。在97 %的相似度下,总共产生了14 360个OTU,各样品测序结果如表2所示。在烟草4个生育期中OTU数量呈先下降后又升高,最终在成熟期又下降的趋势,在现蕾期达到峰值。单作与套作在4个时期相应文库覆盖率在93.9 % ~ 96.6 %,所有样品的稀释曲线趋于光滑(图1),表明这些序列基本上反映了真实环境中的细菌群落结构,但稀释曲线仍不饱和,有些微生物种类尚未检出。
通过8个样品基因组DNA中ITS1的基因测序,得到309 552条原始序列,部分低质量序列被过滤,最终得到有效序列为189 716条。在97 %的相似度下,总共产生了3288个OTU,各样品测序结果如表3所示。由图2可以清晰看出,所有样品的稀释曲线趋于光滑,表明这些序列基本上反映了真实环境中的真菌群落结构,但稀释曲线仍不饱和,有些微生物种类尚未检出。
由表3中的Alpha多样性指标中覆盖度率可知,各样品文库覆盖率在99.2 %~99.7 %,已大致代表了该序列中的所有真菌群落结构。从土样中的香农指数可以看出,套作体系下的香浓指数大部分高于单作中的香农指数,表明套作提高了土壤中生物群落差异性。同时套作中的Chao1指数也普遍大于单作,进一步说明万寿菊与烟草套作提高了根际土壤微生物群落的总体丰度。
2.2 各样品间微生物群落相关性分析
以个样品得到的OTUs数目为依据,对各样品间微生物群落相关性状进行分析,并构建细菌群落花瓣图。根据图3可知,单作与套作在4个不同生育期的8组样品中,有752个OTUs均在8个样品中出现。无论是在单作还是套作体系下,在现蕾期OTUs值均达到最高为140和147。随着植物的生长在不同时期有不一样的OTUs数目,存在一定的差异,但也存在一定的规律,总的来说单作烟草变化幅度比较大,套作的OTUs变化相对稳定。
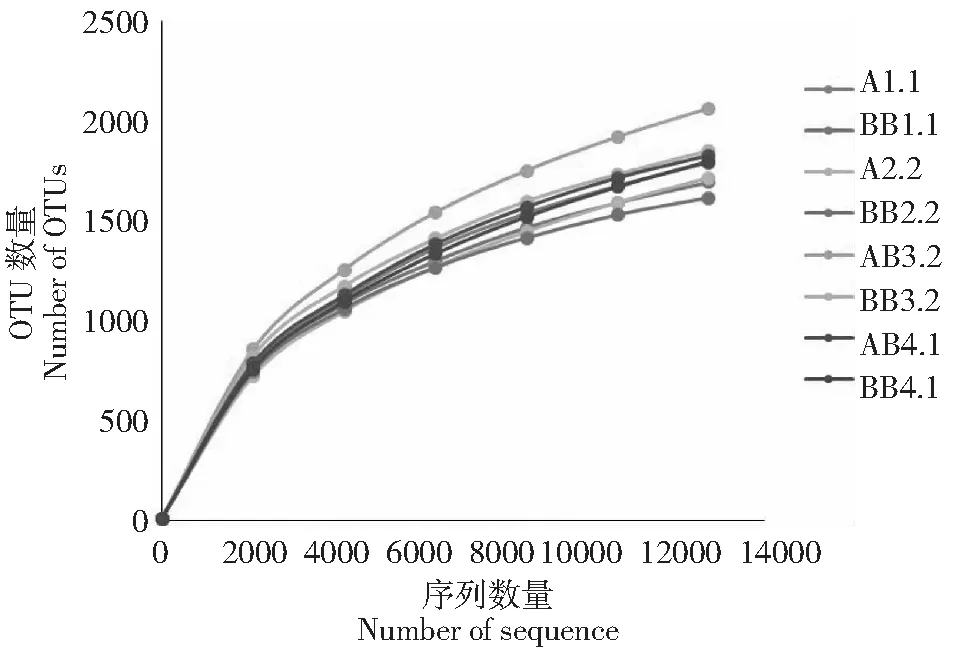
图1 细菌丰度稀释度曲线Fig.1 Rarefaction curves for bacterial
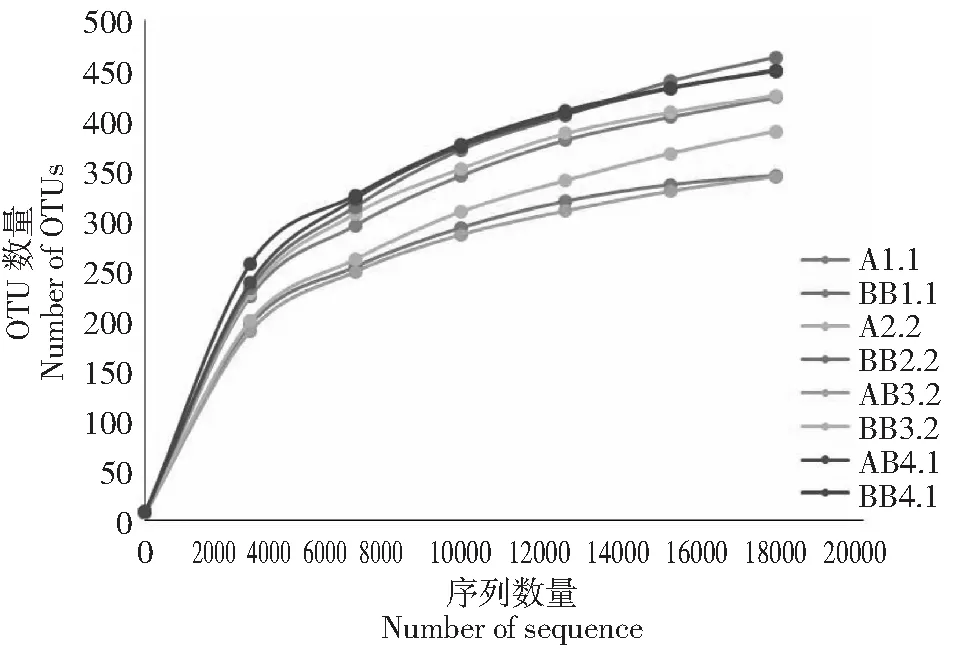
图2 细菌丰度稀释度曲线Fig.2 Rarefaction curves for fungus
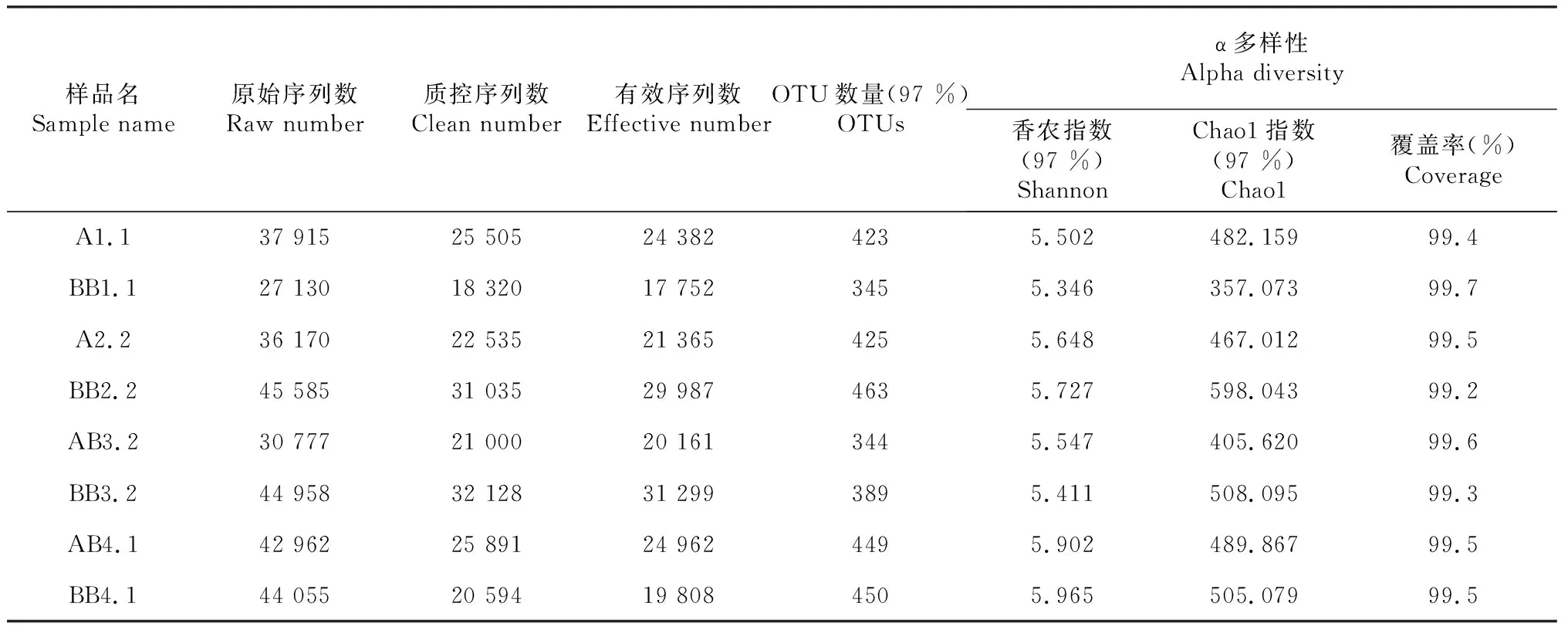
样品名Sample name原始序列数Raw number质控序列数Clean number有效序列数Effective numberOTU数量(97 %)OTUs α多样性Alpha diversity香农指数(97 %)Shannon Chao1指数(97 %)Chao1覆盖率(%)CoverageA1.137 91525 50524 3824235.502482.15999.4BB1.127 13018 32017 7523455.346357.07399.7A2.236 17022 53521 3654255.648467.01299.5BB2.245 58531 03529 9874635.727598.04399.2AB3.230 77721 00020 1613445.547405.62099.6BB3.244 95832 12831 2993895.411508.09599.3AB4.142 96225 89124 9624495.902489.86799.5BB4.144 05520 59419 8084505.965505.07999.5
同样以序列真菌测定中所得的OTUs数目为依据,进行各样品间真菌群落相关性状分析,构建花瓣图。由图4可知,各样品共有的OTUs数目为162,同时无论是单作还是套作每个时期的OTUs数目都大大低于细菌的,说明植物根际微生物群落中细菌是主要优势菌种。不同栽培方式在不同时期表现不一样的OTUs,单作中在成熟期OTUs数目达到峰值24,现蕾期时最小为7,套作体系下成熟期达到峰值21,团棵期最小为6。进一步说明套作改变了不同时期真菌群落多样性分布。
2.3 单作与套作模式烟草土壤微生物多样性的PCA分析
从图5可知,主成分1(PC1)可解释全部土壤样品细菌群落多样性的18.27 %,主成分2(PC2)可解释土壤样品细菌群落的16.04 %,两者总计可解释全部土壤样品的34.31 %。图5表明单作烤烟与万寿菊和烤烟套作两处理在不同时期有不同的细菌群落结构,存在明显的差异;样品A1.1与BB1.1均分布在PC1和PC2的正值区域,但二者存在一定距离,说明在团棵期单作与套作土壤细菌群落存在着一定差异。样品BB2.2,BB3.2和BB4.1均分布在PC2的正值区域,PC1的负值区域,但BB2.2与BB3.2和BB4.1距离较远,差异较大,而BB3.2与BB4.1之间距离较近,两者非常相似;同时样品AB3.2与AB4.1和A2.2均分布在PC2的负值区域,但分处在PC1的正负值区域,且间隔位置较大,AB4.1和A2.2与A1.1间隔位置也较远,进一步说明了随着时间的推移,土壤细菌群落分布在显著变化。总体来说土壤细菌群落结构受环境影响较大,时间变异很大。
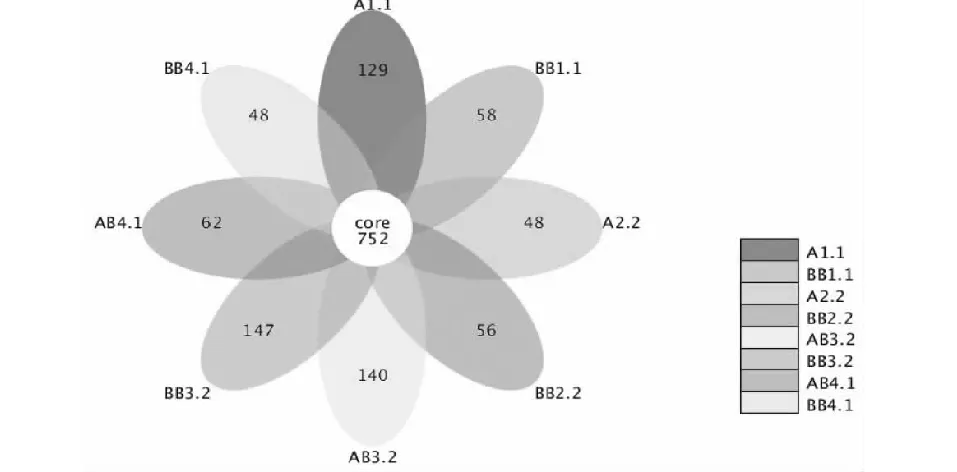
图3 各样品间细菌OTUs花瓣图Fig.3 Flower-figure of OTUs for bacterial sample
对样品真菌PCA主成分的分析结果如图6所示,从图6可以看出,主成分1(PC1)可解释全部土壤样品细菌群落多样性的20.64 %,主成分2(PC2)可解释土壤样品细菌群落的16.77 %,两者总计可解释全部土壤样品的37.41 %。样品A1.1和BB1.1分布在PC1正值区域,PC2的负值区域,同样样品BB4.1和AB4.1均分布在PC2的正值区域,PC1的负值区域但是都存在一定差异。相反BB2.2分布在PC1及PC2的正值区域,而A2.2分布在PC1及PC2的负值区域,BB3.2与AB3.2虽然都分布在PC1的正值区域,但分处在PC2的正负值区域,并且A2.2与BB2.2和AB3.2与BB3.2两两位置间隔距离较远,说明单作与套作在烤烟旺长期和现蕾期真菌群落结构差异非常显著。综合以上所述,烟草根际土壤微生物群落结构受环境影响较大,时间变异较大,在不同生长期表现不同的群落结构,之间差异显著。
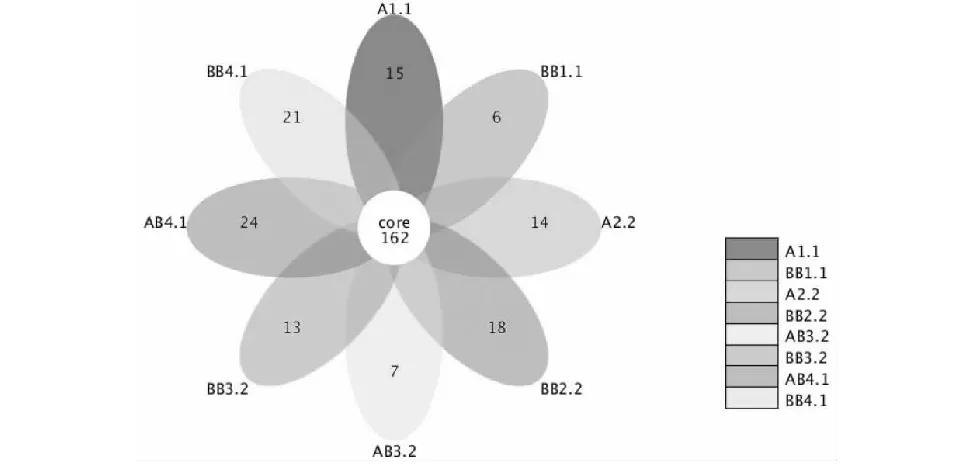
图4 各样品间真菌OTUs花瓣图Fig.4 Flower-figure of OTUs for fungus sample
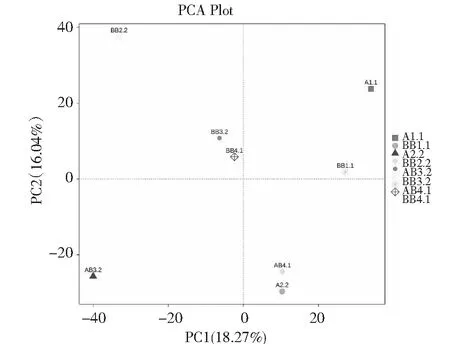
图5 各样品不同时期土壤细菌群落主成分分析Fig.5 PCA analysis of bacterial community in soils at different growth stages
2.4 各样品细菌群落分布特征分析
选取各样品中占各样本细菌群落组成的95 %以上10个门的细菌,进行绘制群落结构分布图图7。由图7可以明显看出无论是在套作还是在单作体系下烟草根际土壤微生物群落变形菌门是主要优势菌门,在烟草整个生长周期中相对百分比都大于25 %,占其细菌群落组成的1/4以上。其次是酸杆菌门、放线菌门、芽单胞菌门和绿弯菌门。在现蕾期单作模式下变形菌门达到最大值(56.2 %),与套作模式下(26.9 %)形成鲜明的对比,就整体而言放线菌门在套作体系下所占比例降低。酸杆菌门在在2种种植模式的团棵期所占比例最小,单作为9.6 %,套作为8.1 %,但单作体系下在现蕾期达到整个生长周期的峰值18.6 %,而套作体系下则是在旺长期达到峰值25.5 %。放线菌门在单作体系中各个生长期所占比例分别是团棵期(21.2 %)、旺长期(14.1 %)、现蕾期(12.3 %)、成熟期(15.6 %),套作增加了其所占比例分别为团棵期(23.5 %)、旺长期(21.2 %)、现蕾期(17.6 %)、成熟期(20.1 %)。对于芽单胞菌门,绿弯菌门和硝化螺旋菌门在单作和套作中波动不大整体趋于稳定。奇古菌门在单作体系下呈现先降低后又增高的趋势,而在套作体系下恰好相反呈现先增高后降低的趋势,厚壁菌门在单作的团棵期达到最大值4.4 %,而后就明显降低,在套作体系中所占比例都低于单作团棵期,并且相对稳定。通过图7还可以看出通过万寿菊与烟草套作处理大大降低拟杆菌门细菌微生物,并使其在各个生长周期中总体趋于稳定状态。
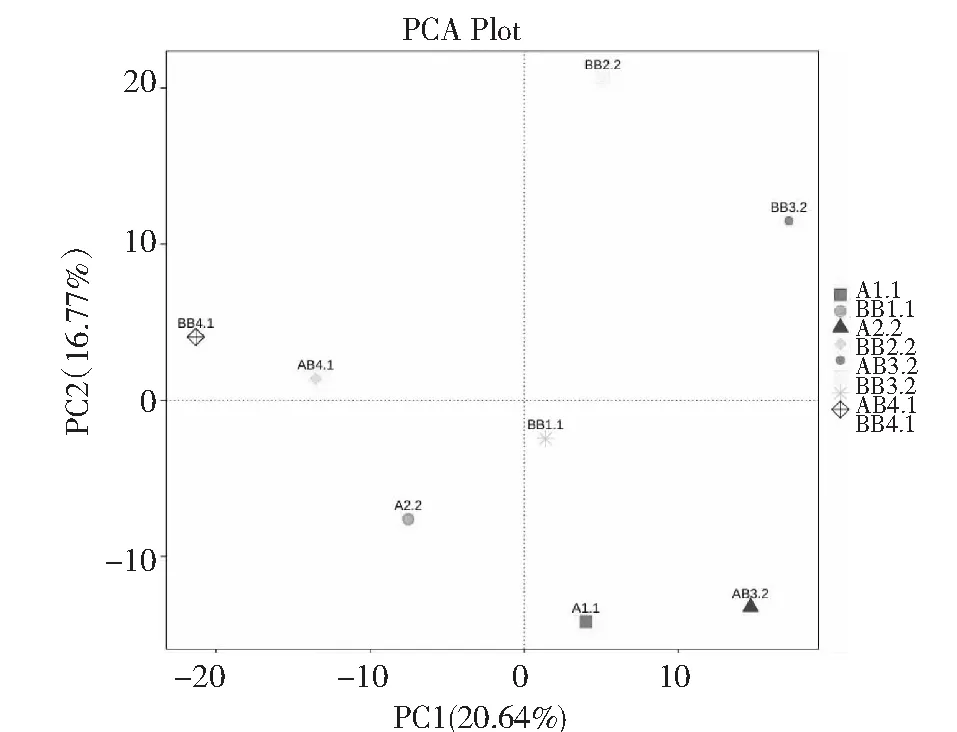
图6 各样品不同时期土壤真菌群落主成分分析Fig.6 PCA analysis of fungus community in soils at different growth stages
从属分类水平上看单作与套作总共检测分类出467个细菌属(图8),细菌种类丰富但仍然还有大量细菌属未分类,各样品间菌属分布差异很大。在单作体系下主要菌属的食碱菌属(Alcanivorax),海杆菌属(Marinobacter)鞘氨醇单胞菌属(Sphingomonas),3个属在团棵期占绝对优势分别是5.65 %、5.45 %、4.49 %,而在套作体系下却相反。
2.5 各样品真菌群落分布特征分析
检测到8个样品共涉及子囊菌门(Ascomycota),接合菌门(Zygomycota),担子菌门(Basidiomycota),壶菌门(Chytridiomycota),Incertae_sedis_Fungi,聚合菌门(Glomeromycota)6个菌门。由图9可知,烟草根际土壤真菌群落组成中首要菌门是子囊菌门,占所有菌门的80 %以上,其次是接合菌门和担子菌门,它们所占比例虽小但均在2.83 %以上。接合菌门主要存在于旺长期而后,通过套作处理可适当降低其比例,单作中担子菌门首先略微下降后又增加,在成熟期又微降至7.29 %,而套作处理的担子菌门主要存在于团棵期,在整个生长周期中呈逐渐下降趋势。
从属分类水平看8个样品总共涉及171个真菌属,各样品间菌种属组成差异显著。从图10可以看出,烟草根际微生物种群中第1位菌属是毛壳属(Chaetomium),第2位菌属是曲霉属(Aspergillus)。在团棵期是根际真菌微生物群落含量最丰富的时期,从图中还可以明显看出单作成熟期Microidium属真菌数量与其它3个时期相比较高为AB4.1(5.32 %),通过套作之后可使其数量降低。套作增加团棵期锥盖伞属(Conocybe)的菌种数量为BB1.1(4.16 %)。
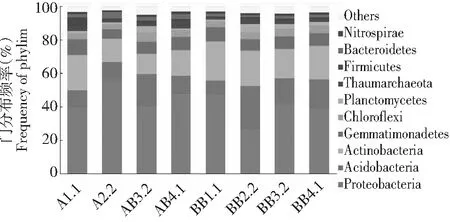
图7 门分类水平各样品土壤细菌菌群结构分布Fig.7 Structure map of soil bacteria on the level of phylum from samples
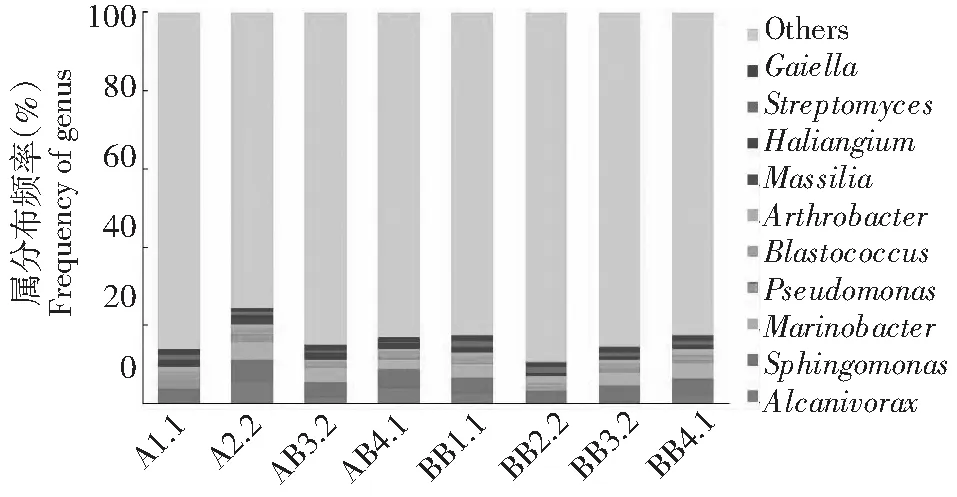
图8 属分类水平各样品土壤细菌菌群结构分布Fig.8 Structure map of soil bacteria on the level of genus from samples
3 讨 论
近几年迅速发展起来的Illumina MiSeq高通量测序技术相比较第一代Sanger测序技术具有通量高,大规模测序成本低,文库覆盖率高等特点[20-22],非常适合用于土壤微生物菌种鉴定。真菌的ITS区域进化较快,种间特异性明显,种内相对保守,且序列长度适中,可以从不太长的序列中获取更多信息,快速准确地鉴定真菌菌种[23-24]。故本研究通过运用Illumina MiSeq第二代测序技术对单作烟草和万寿菊-烟草套作体系下不同生长周期根际8个土样的16S-V4和ITS1变异区进行PCR扩增测序。
本研究通过16S-V4区测序结果多样性分析发现文库覆盖率在93.9 %~96.6 %,表明测序结果很接近真实环境中细菌多样性和物种丰富度。其中香农指数(Shannon)在旺长期先降低又升高,随后在成熟期又下降,在现蕾期达到最大值,表明在整个生长周期中细菌群落物种多样性差异很大。Chao1指数从团棵期到现蕾期在逐渐增加到成熟期降低,而套作处理则是在旺长期以前下降之后增加,这可能是随着植株生长各类细菌不断积累到了成熟期植株组织成熟脱落影响细菌生物活性,而套作处理可能是在旺长期后万寿菊根际分泌一些不利于细菌生长的物质所以降低了其物种种类。对各样品间细菌群落多样性分析发现有752个OTUs是单作与套作两个处理所共有,并且二者独有OTUs均在现蕾期达到峰值。本研究还发现万寿菊-烟草套作体系各生长期根际细菌的变形菌门(Proteobacteria)、酸杆菌门(Acidobacteria)、放线菌门(Actinobacteria)是其三大优势菌门与单作体系下一致,并且主要以变形菌门(Proteobacteria)的α变形菌纲(Alphaproteobacteria)的鞘氨醇单胞菌属(Sphingomonas)和γ变形菌纲(Gammaproteobacteria)的食碱菌属(Alcanivorax),海杆菌属(Marinobacter)为主。
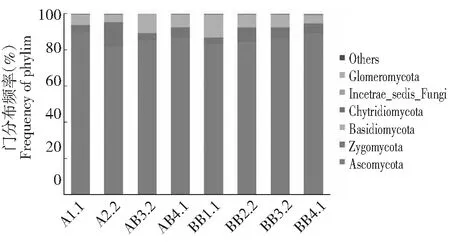
图9 门分类水平各样品土壤真菌菌群结构分布Fig.9 Structure map of soil fungus on the level of phylum from samples
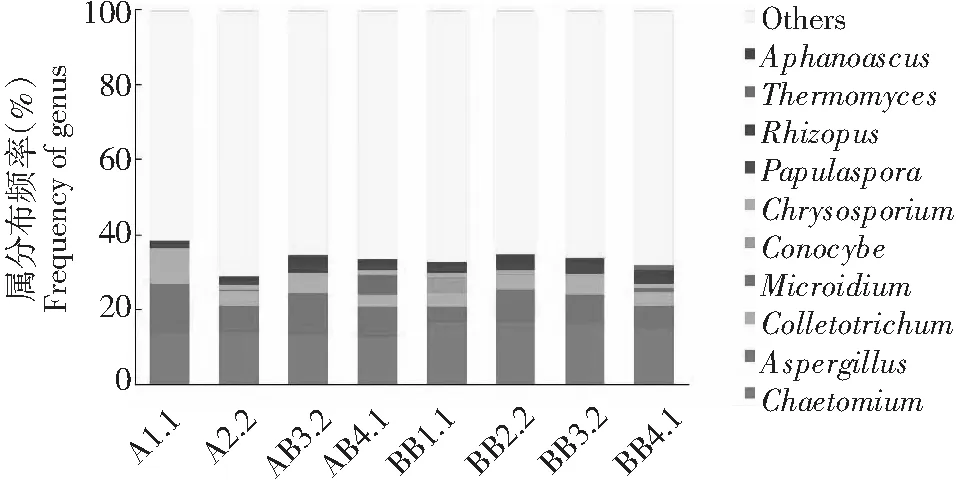
图10 属分类水平各样品土壤真菌菌群结构分布Fig.10 Structure map of soil fungus on the level of genus from samples
在对真菌ITS1区序列测序结果发现每个检测样品文库覆盖率(Coverage)均在99.2 %以上,香农指数(Shannon)在生长周期中都呈现先升高又降低最后再升高的趋势,但与单作相比较套作中Chao1指数几乎都大于单作,表明根际真菌群落在各个生长时期物种多样性不一,可以通过套作处理提高真菌群落物种种类。对各样品间真菌群落多样性分析发现有162个OTUs是两处理所共有的,二者独有OTUs均在成熟期达到峰值。在通过对各个生长期根际土样真菌群落组成中发现,在烟草整个生长周期里根际真菌群落3大优势菌门分别是子囊菌门(Ascomycota)、接合菌门(Zygomycota)和担子菌门(Basidiomycota),其中主要以子囊菌门(Ascomycota)的粪壳菌纲(Sordariomycetes)中的毛壳属(Chaetomium)及炭疽菌属(Colletotrichum)和散囊菌纲(Eurotiomycetes)中的曲霉属(Aspergillus)为主。
4 结 论
通过对细菌16S-V4区和真菌ITS1区的测序注释分析全面了解万寿菊-烟草套作体系下不同生育期微生物种类变化情况,套作虽在不同生育期改变植烟环境土壤微生物多样性及物种数量,但不能完全改变植烟土壤环境微生物种类组成。本研究检测到很大比例的未分类菌群,说明还有大量未知微生物资源有待发掘。
参考文献:
[1]时 鹏, 张继光, 王正旭, 等. 烟草连作障碍的症状·机理及防治措施[J]. 安徽农业科学, 2011, 39(1): 120-122, 124.
[2]张继光, 申国明, 张久权, 等. 烟草连作障碍研究进展[J]. 中国烟草科学, 2011, 32(3): 95-99.
[3]阮维斌, 王敬国, 张福锁, 等. 根际微生态系统理论在连作障碍中的应用[J]. 中国农业科技导报, 1999, 1(4): 53-58.
[4]田生辉, 赵彦玲, 纪永存, 等. 连作对设施黄瓜产量和品质的影响[J]. 安徽农学通报, 2014, 20(18): 69-69.
[5]岳冰冰, 李 鑫, 张会慧, 等. 连作对黑龙江烤烟土壤微生物功能多样性的影响[J]. 土壤, 2013, 45(1): 116-119.
[6]冯俊喜, 王树声, 石 屹, 等. 山东烟区不同种植模式土壤微生物群落特征研究[J]. 中国烟草科学, 2011, 32(2): 38-42.
[7]刘丽芳, 唐世凯, 熊俊芬, 等. 烤烟间套作草木樨和甘薯对烟叶含钾量及烟草病毒病的影响[J]. 中国农学通报, 2006, 22(8): 238-241.
[8]涂 勇, 杨文钰, 刘卫国, 等. 大豆与烤烟不同套作年限对根际土壤微生物数量的影响[J]. 作物学报, 2015, 41(5): 733-742.
[9]阳显斌, 李廷轩, 张锡洲, 等. 烟蒜轮作与套作对土壤农化性状及烤烟产量的影响[J]. 核农学报, 2015, 29(5): 980-985.
[10]田 峰, 蒋才军, 陈治锋, 等. 烟薯不同套作时期对烤烟影响及综合效应的研究初报[J]. 湖南烟草, 2009, S1: 238-244.
[11]贾兰虹, 王 涛, 霍 祥, 等. 万寿菊圆葱生态协调互作丰产栽培技术研究[J]. 黑龙江农业科学, 2006(4): 59-62.
[12]魏环宇, 管丽蓉, 王 扬, 等. 万寿菊当归多样性种植对土壤真菌多样性的影响[J]. 植物保护, 2015, 41(5): 69-74, 93.
[13]孙建波, 邹良平, 李文彬, 等. 香蕉不同生育期根际土壤细菌群落变化研究[J]. 热带作物学报, 2016, 37(6): 1168-1171.
[14]Caporaso J G, Lauber C L, Walters W A, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample[J]. Proceedings of the National Academy of Sciences of the United States of America, 2011, 108(Suppl 1): 4516-4522.
[15]Hess M, Sczyrba A, Egan R, et al. Metagenomic discovery of biomass-degrading genes and genomes from cow rumen[J]. Science, 2011, 331(616): 463-467.
[16]Edgar R C, Haas B J, Clemente J C, et al. UCHIME improves sensitivity and speed of chimera detection[J]. Bioinformatics, 2011, 27(16): 2194-2200.
[17]Edgar R C. UPARSE: highly accurate OTU sequences from microbial amplicon reads[J]. Nature Methods, 2013, 10(10): 996-998.
[18]Wang Q, Garrity G M, Tiedje J M, et al. Naive bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy[J]. Applied and Environmental Microbiology, 2007, 73(16): 5261-5267.
[19]Desantis T Z, Hugenholtz P, Larsen N, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB[J]. Applied and Environmental Microbiology, 2006, 72(7): 5069-5072.
[20]陈泽斌, 李 冰, 王定康,等. Illumina MiSeq高通量测序分析核桃内生细菌多样性[J]. 江苏农业学报, 2015, 31(5): 1129-1133.
[21]陈泽斌, 李 冰, 王定康, 等. 应用Illumina MiSeq高通量测序技术分析玉米内生细菌多样性[J]. 现代食品科技, 2016, 32(2): 113-120.
[22]陈泽斌, 李 冰, 王定康, 等. 白芨内生细菌组成及多样性分析[J]. 南方农业学报, 2016, 47(2): 227-233.
[23]曹小迎, 蒋继宏, 孙 勇, 等. 刺革菌科4种药用真菌的ITS区序列分析[J]. 中草药, 2007, 38(2): 261-264.
[24]燕 勇, 李卫平, 高雯洁, 等. rDNA-ITS序列分析在真菌鉴定中的应用[J]. 中国卫生检验杂志, 2008, 18(10): 1958-1961.
猜你喜欢
河北农业(2023年10期)2023-11-28 09:23:34
广西林业科学(2022年2期)2022-05-09 07:49:54
云南农业(2021年9期)2021-09-24 11:56:56
四川蚕业(2020年3期)2020-07-16 08:09:44
山东农业科学(2019年11期)2019-12-24 01:11:27
作文新天地(2019年25期)2019-11-27 21:35:44
江苏农业科学(2019年11期)2019-07-22 01:32:45
干旱地区农业研究(2017年5期)2017-12-18 06:18:18
新疆农垦科技(2016年2期)2016-08-21 13:50:18
现代农业(2016年5期)2016-02-28 18:42:34
