
左乙拉西坦的制备方法
2018-05-30 10:36赵会,阎欢,吴静
沈阳化工大学学报 2018年1期
赵 会, 阎 欢, 吴 静
(东北制药集团股份有限公司, 辽宁 沈阳 110027)
左乙拉西坦是由比利时UCB公司研究开发的抗癫痫新药,为吡咯烷酮衍生物,2000年4月获FDA批准,目前已在多个国家上市,2007年初在中国上市,主要用于治疗局限性及继发性全身性癫痫[1].其具有选择性、保护局限性和原发全身性癫痫的独特作用,并具有预防癫痫发作的独特性能,且不影响其他抗癫痫药物在体内的代谢.该药药效明确,在美国药典第33~36版和欧洲药典第7~8版均有收载.
自比利时UCB公司开发左乙拉西坦以来,左乙拉西坦的合成研究引起了世界的重视,纷纷寻找更经济、更适用、更环保、转化率更高的化学合成路线.以色列TEVA公司申请了左乙拉西坦制备的专利.许多文献[2-7]专门介绍了左乙拉西坦的化学合成方法.左乙拉西坦化学名为(s)-α-乙基-2-氧代-1-吡咯烷基乙酰胺,是唯一被证实具有与突触前神经末梢内突触小泡蛋白SV2A结合的抗癫痫药物,其易溶于水,口服后经胃肠道快速且完全吸收,给药后约1~3 h达血药峰值,食物不影响其吸收,其与血浆蛋白结合率低于10 %,不经过肝脏代谢,在血液中由酶水解为无活性代谢物,随尿液排出,半衰期约为6~8 h,对老年人和肾功能受损病人半衰期较长.
综上所述,左乙拉西坦是一个毒副作用小、疗效好的极具开发价值的广谱抗癫痫药,因此,研究和开发一条简便且适合工业化生产的合成路线十分必要.
1 实验部分
1.1 主要仪器和药品
MSU125P-100DU电子天平,北京慧龙环科环境仪器有限公司;Waters 2695-12489 UV-VIS检测器,沃特世科技有限公司;NICOLET iS10傅里叶变换红外光谱仪,赛默飞世尔科技公司;Waters ACQUITY UPLC-Xevo TQ超高效液相色谱-质谱联用仪,沃特世科技上海有限公司;microTOF-Q质谱联用仪,美国布鲁克·道尔顿公司;PE Lambda 35 UV/VIS分光光度计,美国布鲁克·道尔顿公司;ARX-300型核磁共振仪,瑞士 Bruker公司;METTLER TOLEDO DSC1差示扫描量热仪,梅特勒一托利多仪器上海有限公司.
S-2-氨基丁酰胺盐酸盐,苏州天马精细化学品有限公司生产;4-氯丁酰氯,连云港群盛化工有限公司生产;四丁基溴化铵及其他试剂,东北制药集团股份有限公司生产.
1.2 工艺路线
工艺路线如下:
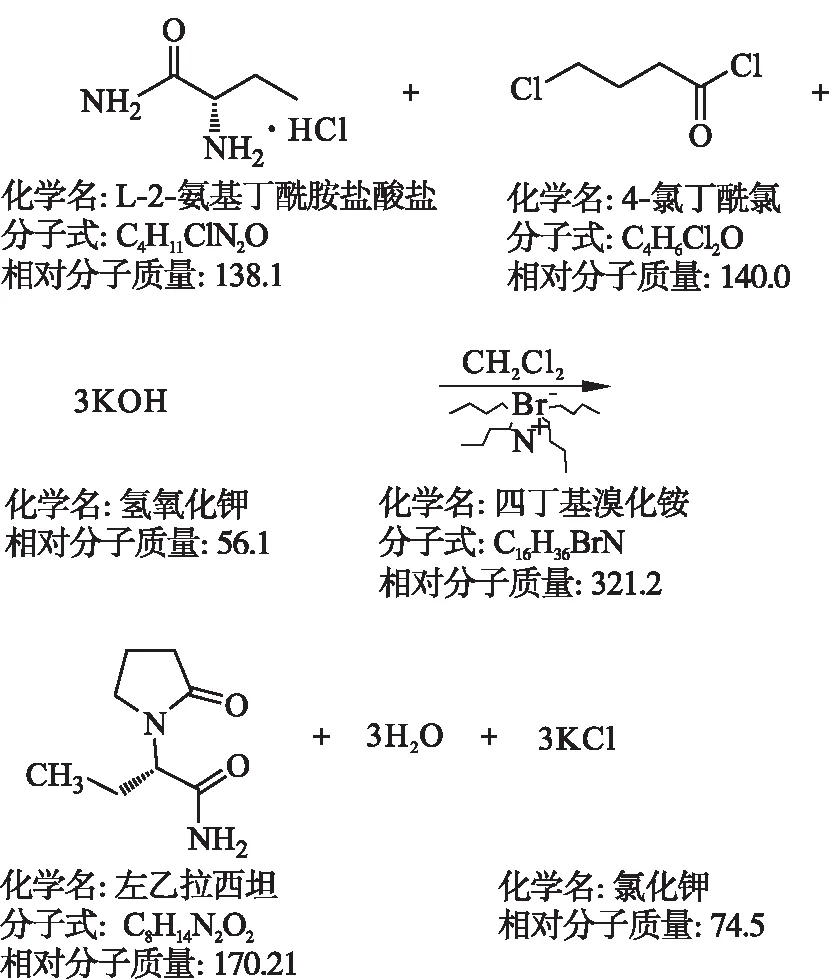
1.3 合成
1.3.1 环合反应
取1 000 mL三颈瓶加入S-2-氨基丁酰胺盐酸盐42 g,加入二氯甲烷400 mL,降温至-10 ℃,加入四丁基溴化铵19.4 g,搅拌10 min,加入氢氧化钾34 g,搅拌15 min,滴加4-氯代丁酰氯21.6 g和二氯甲烷40 mL溶液,45 min滴加完毕,-8 ℃搅拌保温45 min;加入氢氧化钾34 g,-8 ℃搅拌15 min,滴加4-氯代丁酰氯12.8 g和二氯甲烷溶液40 mL,-8 ℃下滴加30 min,-8 ℃下保温45 min;加入氢氧化钾17 g,-8 ℃搅拌15 min,滴加4-氯代丁酰氯12.8 g和二氯甲烷溶液 40 mL,30 min滴加完毕,-8 ℃保温45 min;整个反应不超过-6 ℃.反应液在-2 ℃下保温 5 h,加入8.6 g氢氧化钾保温2 h,过滤.滤液使用醋酸调节pH值,使之为7.0~7.5,加入60 g无水硫酸钠干燥过夜.干燥完毕后过滤,滤液减压浓缩至干,加入50 mL乙酸乙酯分两次带出残留的二氯甲烷.加入240 mL乙酸乙酯,5~10 ℃结晶5 h,5~10 ℃过滤,少量乙酸乙酯洗涤,得粗品左乙拉西坦,收率范围为72.0 %~77.0 %(摩尔分数),含量≥90.0 %(HPLC,质量分数).
1.3.2 精制
取粗品45~48 g,加入丙酮100 mL,纯化水1.5 g,60 ℃回流至完全溶解后5 ℃条件下析晶过夜.析晶结束后抽滤,用少量丙酮洗涤,得一精品30~33 g.取一精品,加入丙酮90 mL,纯化水1.5 g,60 ℃下加热回流至晶体完全溶解后7 ℃析晶5 h,析晶完毕后过滤得成品左乙拉西坦,收率55 %~60 %(摩尔分数),含量≥97.0 %(HPLC,质量分数).
2 结果与讨论
2.1 谱图分析
由高分辨质谱给出本品的元素组成为C8H14N2O2,与左乙拉西坦(化学结构见图1)的分子式一致;IR吸收光谱显示伯酰胺中氨基(—NH2)的对称和不对称伸缩振动(~3 360、~3 192 cm-1)、酰胺上羰基(—C==O)的伸缩振动(~1 680 cm-1)、甲基和乙基的对称和不对称伸缩振动(~2 991、~2 939 cm-1)以及内酰胺环的伸缩振动和变角振动(~1 295、~636 cm-1),均为左乙拉西坦的主要官能团在红外光谱中的表现,自制品的红外光谱吸收峰位置、峰形、强弱均与对照品一致;UV吸收图谱中,仅存在n—π*跃迁,应由酰胺的羰基结构引起,与左乙拉西坦UV特征一致,并且自制品与对照品在四种介质中的UV图谱及吸收系数基本一致;在1H-NMR谱数据中,自制品与对照品均呈现10组谱线,对应于14个氢原子,与左乙拉西坦化学结构中质子的类型数和个数一致;根据各种氢的化学位移、裂分情况和对应的质子数分别进行归属,与左乙拉西坦的化学结构相符,自制品与对照品1H-NMR谱一致;在13C-NMR谱中,自制品与对照品均呈现8组谱线,对应于8种碳原子,与左乙拉西坦化学结构中碳原子的类型数和个数一致;根据各种碳的化学位移分别进行归属,与左乙拉西坦的化学结构相符,自制品与对照品13C-NMR谱一致;液质联用(MS-HPLC)总离子流图自制品与对照品保留时间一致(均为1.8 min),一级MS显示本品相对分子质量为170,与左乙拉西坦理论相对分子质量一致,(M+H)+峰的二级MS裂解碎片离子与左乙拉西坦的可能裂解途径相符,自制品与对照品的一级、二级MS图谱均一致;DSC谱图中自制品和对照品均在117 ℃左右呈一个尖锐吸热峰,与左乙拉西坦的熔点(115~119 ℃)一致;未见其他峰,说明样品为无水无溶剂的纯物质;自制品与对照品的DSC峰位、峰形及峰数均一致,说明自制品与对照品热力学行为一致.
1H-NMR和13C-NMR的氢和碳的化学位移解析和归属见表1、表2.
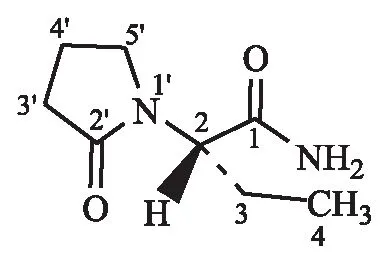
图1 左乙拉西坦化学结构Fig.1 Levetiracetam chemical structure表1 1H-NMR的氢和碳的化学位移解析和归属Table 1 Chemical shift and attribution of 1H-NMR hydrogen and carbon
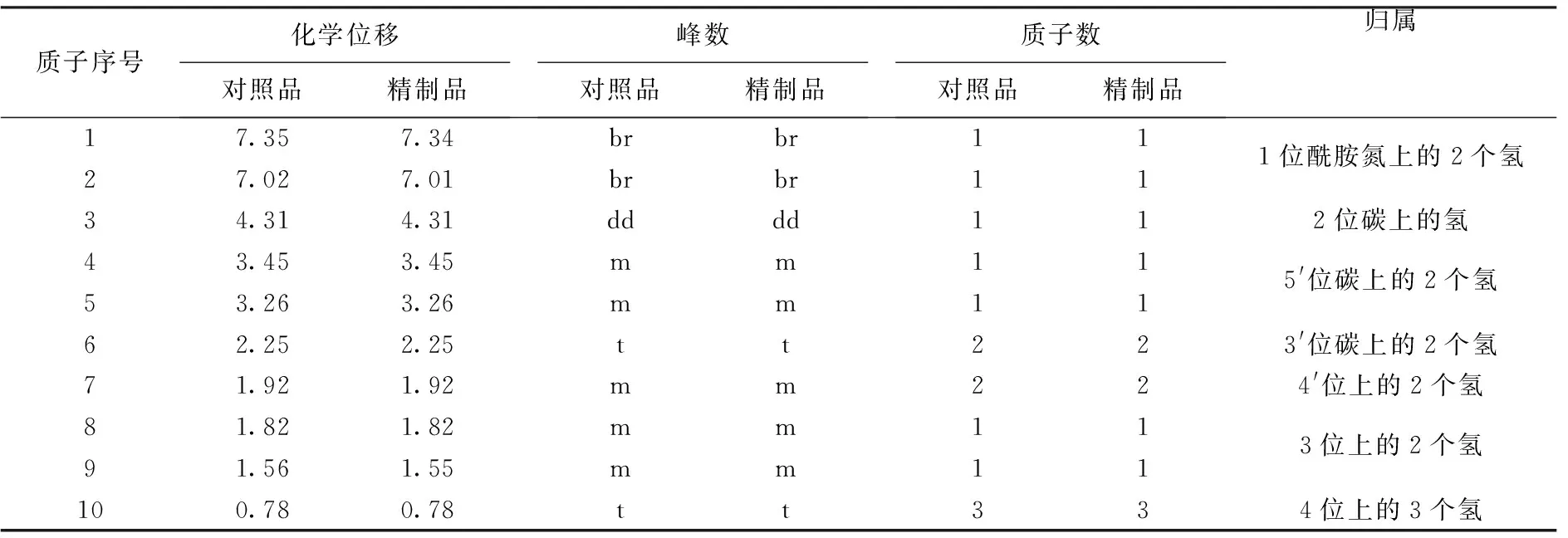
质子序号化学位移对照品精制品峰数对照品精制品质子数对照品精制品归属17.357.34brbr111位酰胺氮上的2个氢27.027.01brbr1134.314.31dddd112位碳上的氢43.453.45mm115'位碳上的2个氢53.263.26mm1162.252.25tt223'位碳上的2个氢71.921.92m m 224'位上的2个氢81.821.82mm113位上的2个氢91.561.55mm11100.780.78tt334位上的3个氢

表2 13C-NMR的氢和碳的化学位移解析和归属Table 2 Chemical shift resolution and home of 13C-NMR hydrogen and carbon
综上所述,左乙拉西坦自制品所有光谱数据与左乙拉西坦对照品均一致,且与左乙拉西坦结构相吻合,可以确定自制品为左乙拉西坦.
2.2 讨 论
参考文献[8],以a-吡咯烷酮为起始原料,经成盐、酯化、水解、拆分、氨化得到左乙拉西坦,总收率37.8 %(摩尔分数)左右,此合成路线原料易得,但由于步骤长、收率低、成本高等因素,不适合大生产;另有文献[1]以L-2-氨基丁酸与二氯亚砜反应得到L-2-氨基丁酰氯,再与甲醇酯化得到L-2-氨基丁酸甲酯,与4-溴丁酸甲酯、碳酸钠在水中回流反应得到2(S)-[3-(乙氧羰基)丙氨基]丁酸甲酯,然后再在2-羟基吡啶中回流得到2-(S)-(2-氧代吡咯烷基)丁酸甲酯,最后氨解得到左乙拉西坦,收率46 %(摩尔分数)左右.该路线由于在起始原料中已经引入了手性源,可以避开手性拆分,但是由于步骤长、4-溴丁酸甲酯价格高、以及二氯亚砜严重腐蚀设备等因素,该路线不适合大生产;又有文献[9]以2-吡咯烷酮与氧代丁酸在甲苯中回流得到2-(2-氧代吡咯烷-1-基)-2-丁烯酸,在四氢呋喃中与五氯化磷反应得2-(2-氧代吡咯烷-1-基)-2-丁酰氯,再通入氨气得到相应的酰胺,最后用手性催化剂在5×101.325 kPa下进行不对称氢化得到左乙拉西坦.该合成路线通过立体选择性还原的方法解决了手性问题,但是由于Rh(Ⅰ)*或Ru(Ⅱ)*手性催化剂来源受限,并且价格昂贵,综合各因素该路线目前不适合大生产.
本合成路线与浙江大学硕士论文[4]中报道的合成方法三的工艺路线类似,文献中以S-2-氨基丁酸为原料,而本实验直接以S-2-氨基丁酰胺盐酸盐为原料合成左乙拉西坦;文献中以甲苯为析晶溶剂,本实验以乙酸乙酯代替毒性大的甲苯,得到的产品不仅晶型好,而且收率高;文献直接采用丙酮重结晶得到终产品,本实验采用丙酮和水先加热回流后再进行重结晶两次,产品收率高,质量好,有利于工业化生产.
3 结 论
研究表明左乙拉西坦治疗癫痫具有较好疗效,是一种广谱、安全、耐受性好的新型抗癫痫药物,其在临床上的应用也越来越受到人们的重视,因此研究和开发出一条适合工业化生产且操作简便的合成方法是十分必要的.本合成方法通过文献检索以及多次实验研究,以国内有售、质量可控的S-2-氨基丁酰胺盐酸盐和4-氯丁酰氯为起始原料,经环合反应得环合物(左乙拉西坦粗品),对粗品精制得到终产品左乙拉西坦.该方法合成步骤少,工艺简单,原料易得,产品质量可控,工艺稳定可靠,适合工业化生产.
:
[1] 罗湘冀.左乙拉西坦的合成[J].药学进展,2004,28(9):413-417.
[2] 王飞龙,范兴山,赵海桥.左乙拉西坦的合成工艺改进[J].中国化工贸易,2013(8):382.
[3] 薛娜,周春节,张恺,等.左乙拉西坦的合成[J].河北医科大学学报,2013,34(9):1048-1050.
[4] 楼磊.左乙拉西坦合成工艺的研究[D].浙江:浙江大学,2012:10-19.
[5] 李晓东.抗癫痫药左乙拉西坦(Levetiracetam)[J].国外医药(合成药.生化药.制剂分册),2001,(2):125.
[6] 汪洪湖,韦亚锋,陈文婕.左乙拉西坦合成工艺优化研究[J].安徽医药,2011,15(6):681-683.
[7] 李袆亮.抗癫痫新药左乙拉西坦的工艺研究[D].北京:中国协和医科大学,2002:15-24.
[8] 刘跃金,陈道鹏,梁良,等.左乙拉西坦及其衍生物的合成[J].中国新药杂志,2007,16(11):860-864.
[9] DIFFERDING E,KENDA B,LALLEMAND B,et al.2-Oxo-1-Pyrrolidine Derivatives,Process for Preparing Them and Their Uses:US,20100222576A[P].2010-09-02.http://www.patentsencyclopedia.com/app/20100222576
猜你喜欢
化工时刊(2022年6期)2022-09-01
昆明医科大学学报(2021年10期)2021-12-02
中国医院用药评价与分析(2021年2期)2021-04-23
中华养生保健(2020年3期)2020-11-16
中华养生保健(2020年1期)2020-11-16
中外医疗(2019年16期)2019-09-02
中国社区医师(2019年17期)2019-08-21
分析化学(2019年3期)2019-03-30
医药前沿(2019年11期)2019-01-05
中国医药指南(2017年3期)2017-11-13
