
RAGE阻断剂FPS-ZM1对心肌缺血再灌注损伤的作用研究
2018-05-29 08:47
实用老年医学 2018年5期
针对急性心肌梗死病人,实施及早、有效的再灌注治疗如溶栓或者经皮冠脉介入,是减少梗死面积、改善临床预后的治疗措施[1]。然而,恢复血流的同时有可能带来局部细胞凋亡、炎症或者氧化负荷,导致不可逆细胞损伤,这种现象即心肌缺血再灌注损伤(myocardial ischemia reperfusion injury,MIRI)。MIRI可引起心脏功能不全包括心律失常、微血管损伤和细胞死亡。损伤机制包括大量活性氧(ROS)激活、钙超载、促炎性因子释放、凋亡发生、中性粒细胞浸润、内皮细胞功能不全[2-3]。如何减轻和避免MIRI成为再灌注治疗的重要内容。大量实验表明,远端缺血预适应(remote ischemic percondition-ing,RIPerC)[4]和缺血后适应(remote ischemic postconditioning,RIPostC)[5]干预措施能够减轻缺血再灌注损伤。
最近研究把晚期糖基化终末产物受体(receptor for advanced glycation end products,RAGE)放在介导MIRI的信号通路的中心位置。RAGE是一种多配体的膜受体,可表达于大多数组织和一系列细胞中,在炎症发展过程中起到重要作用,与配体高迁移率族蛋白B1(high-mobility group box 1,HMGB1)、晚期糖基化终末产物(advanced glycation end products,AGEs)、β 淀粉样蛋白(β-amyloid,Aβ) 等结合后,可启动多条信号通路,引起细胞内氧化应激和炎症反应,细胞功能紊乱,导致糖尿病、动脉粥样硬化、MIRI等疾病的发生[6]。FPS-ZM1(FZM1)是RAGE的特异性阻断剂[7-8],本文将着重探究FZM1在小鼠MIRI模型中的具体作用及其信号通路。
1 材料与方法
1.1 材料 7~9周龄健康雄性C57BL/B6小鼠(南京青龙山动物中心)32只,体质量20~25 g,无特定病原体(specific pathogen free, SPF)级。使用仪器和试剂:小鼠呼吸机,小鼠心超仪(Vevo770 imaging system,加拿大),显微手术器械,自制小鼠气管插管,伊文氏蓝(Evans blue,Sigma公司,美国),BCA蛋白定量,试剂盒(碧云天公司,上海),抗RAGE、HMGB1、P-ERK1/2(cell signaling公司,美国),抗ERK1/2抗体、GAPDH抗体(Bioworld 公司)。
1.2 方法
1.2.1 小鼠心肌缺血再灌注模型的建立:小鼠腹腔内注射1%戊巴比妥,通过自制小鼠气管导管成功连接小鼠呼吸机(呼吸频率90~100次/min),核心体温保持在37 ℃左右。在第3、4肋间开胸,暴露出左心耳及左心室,在左心耳下缘2 mm处用7-0的带线缝合针横穿于冠状动脉前降支并结扎(打一活结),观察左心室前壁颜色由红色逐渐变白色并且室壁运动减弱表明结扎成功,45 min之后打开活结实现缺血后的再灌注,封胸。
1.2.2 实验分组与干预:小鼠随机分为4组,即假手术组(sham组,连续腹腔注射1周生理盐术,开胸,不结扎),FPS-ZM1对照组(FZM1组,连续腹腔注射1周1 mg/kg FPS-ZM1,开胸,不结扎),缺血再灌注组(I/R组,腹腔注射生理盐水,结扎左冠状动脉45 min),FPS-ZM1干预组(FZM1+I/R组,手术前腹腔注射FPS-ZM1,结扎左冠状动脉45 min)。
1.2.3 血流动力学测定:再灌注1周后,小鼠给予1%戊巴比妥钠,通过M型超声(Vevo770 imaging system,加拿大)测定左室射血分数(left venricular ejection fraction,LVEF)、左室缩短分数(left venricular fractional shortening,LVFS)。
1.2.4 苏木精-伊红(HE)染色:小鼠超声检测后,取出心脏组织放入4%的甲醛溶液中24 h,使用切片机切取5 μm左右的组织, HE染色后在光镜下观察心肌超微结构变化。
1.2.5 Western blot:再灌注24 h后取出心脏,加入组织裂解液,用BCA蛋白定量试剂盒(碧云天)测定总蛋白,加入一定比例的蛋白上样缓冲液100 ℃煮5 min, 取30 μg蛋白进行10%SDS-聚丙烯酰胺凝胶电泳,转移至PVDF膜上,5%脱脂牛奶封闭2 h,分别加入GAPDH(1∶10000),抗RAGE(1∶1000),抗HMGB1(1∶1000),抗P-ERK(1∶1000),抗ERK(1∶500),4 ℃过夜,洗膜3次,每次10 min,加入含有辣根过氧化物酶标记的二抗(1∶10000),室温2 h,洗膜,最后ECL,孵育,曝光。
1.3 统计学处理 所有数据应用SPSS 19.0统计软件进行数据分析,结果以均数±标准差表示,组间比较采用单因素方差分析(one-way ANOVA),两两比较采用LSD-t检验,P<0.05表示差异有统计学意义。
2 结果
2.1 各组血流动力学指标的变化 与sham组和FZM1组相比,I/R组小鼠心脏收缩功能指标(LVEF、LVFS)明显降低;与I/R组相比,FZM1+I/R组LVEF及LVFS均有所上升,说明FZM1能使MIRI小鼠的心脏收缩功能得到一定恢复(P<0.05)。见表1。
表1 各组小鼠LVEF、LVFS比较%,n=8)
注: 与 sham组比较,*P<0.05;与I/R 组比较,△P<0.05;与FIM1组比较,#P<0.05
2.2 HE染色 与sham组相比,I/R 心肌纤维结构排列紊乱,发生细胞坏死,炎细胞浸润明显,与I/R组相比,FZM1+I/R组的心肌坏死灶面积有一定缩小,炎性细胞浸润明显减少。见图1。
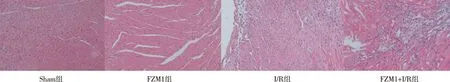
图1 各组心脏组织的镜下超微结构
2.3 蛋白表达比较 与sham组相比,I/R组RAGE及HMGB1表达量升高(P<0.05);与I/R组相比,FZM1+I/R组RAGE及HGMB1表达量显著减少(P<0.05),而P-ERK/ERK表达量显著上升(P<0.01)。见图2,3。
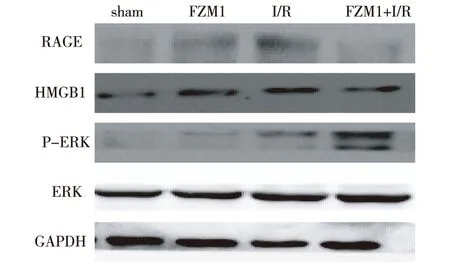
图2 各组蛋白表达的Western blot
3 讨论
再灌注治疗,尤其是溶栓和抗血小板治疗能够显著改善急性心肌梗死病人的临床预后,然而再灌注治疗之前的缺血损害[9],可以触发ROS系统的激活,导致心肌细胞的功能障碍,细胞死亡及炎症反应[10]。最近几年,不少学者已经关注于研制出有效的药物干预措施去加强缺血再灌注损伤的心肌耐受程度。
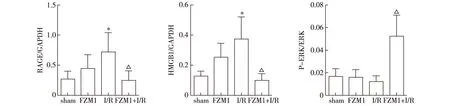
注:与 sham组比较,*P<0.05; 与I/R 组比较, △P<0.05图3 心肌组织RAGE、HMGB1、P-ERK/ERK蛋白表达变化
之前研究已经指出,RAGE及其配体已经参与到MIRI, AGEs和其他的促炎性因子以及组织损伤性配体与RAGE结合后,能够激活许多重要的下游通路。部分研究报道可溶性晚期糖基化终末产物受体(soluble advanced glycation end products,sRAGE)干预或者基因敲除RAGE之后,能够保护再灌注心脏面免受损伤和功能不全,改善心脏功能和代谢[9]。本研究通过药物FZM1干预缺血再灌注小鼠成功阻断RAGE(可能是FZM1直接成功阻断RAGE表达),小鼠心脏的HE染色结果显示,心肌细胞发生炎症、坏死、水肿的程度明显减轻,心脏LVEF由(48.23±0.74)%升高到(52.59±1.05)%,LVFS由(23.74±0.29)%升高到(26.37±0.29)%。
HMGB1是一种非染色体核蛋白,参与稳定核小体和调控基因表达,能够从激活的免疫细胞中主动释放出来,或者从坏死和凋亡细胞中被动释放出[11]。研究证实,细胞外的HMGB1是一种潜在的促炎性因子,能够促进中性粒细胞聚集并激活单核细胞释放更多的炎性因子,在MIRI中有助于放大炎性反应[12-14]。本实验结果显示I/R组的HMGB1蛋白表达量较sham组明显增加,且HE图片上显示I/R组心肌发生明显炎症,说明手术干预使心肌细胞发生了坏死或者凋亡等应激之后,释放了许多促炎性因子,促使心肌组织发生炎症反应,而HMGB1引导的炎症反应主要是通过激活RAGE来实现的[15]。在慢性间歇缺氧(chronic intermittent hypoxia,CIH)动物模型中,HMGB1可能是sRAGE的主要作用靶点,腹腔注射sRAGE(相当于阻断RAGE表达)能在一定程度上降低由CIH引起的RAGE及HMGB1释放量,sRAGE可能是通过下调HMGB1发挥抗炎作用的[16]。根据以上研究结果,本实验Western blot结果可解释为两方面原因:其一,FZM1(阻断RAGE)干预可以直接下调由I/R引起的HMGB1蛋白表达而发挥保护作用。其二,根据HE染色结果,可判断出FZM1能够降低由I/R导致的发生坏死或者凋亡的心肌细胞数目比例,进而减少由心脏组织中坏死细胞释放的HMGB1总量。另有报道称在缺血性心脏病中,血管紧张素转化酶抑制剂(angiotensin converting enzyme inhibitors,ACEI)或者血管紧张素Ⅱ受体拮抗剂能够上调血管紧张素转换酶-2(angiotensin converting enzyme-2,ACE2)的水平,通过阻断HMGB1下游炎症信号的传递起到心脏保护作用,能够使心脏收缩功能指标LVEF值提高21%[17]。本研究中FZM1对缺血再灌注的保护作用可能就是通过阻断HMGB1-RAGE信号轴来抑制HMGB1下游信号的传递,而使得心脏收缩功能增强。HMGB1可以通过与其特异性受体包括RAGE、toll样受体-2(toll-like receptor-2,TLR-2)、TLR-4结合发挥促炎作用[18],配体与受体的结合能够激活核转录因子(NF-kB)释放,或者通过影响丝 裂 原 活 化 蛋 白 激 酶 (mitogen activated protein kinase, MAPK) 信号[19],最终导致大量炎性因子的表达增加和释放。由此可见,HMGB1与RAGE信号轴是一条重要的炎性信号通路,可以介导MIRI的发生发展。MAPK是蛋白酶超家族,由细胞外信号调控酶和细胞外信号调节蛋白激酶(extracellular signal regulated protein kinase, ERK1/2) 、c-Jun N-末端激酶(c-Jun N-terminal kinase,JNK)、p38 组成。MAPK中的p38和JNK能够引起炎症、细胞凋亡[20],而ERK1/2的作用比较复杂,有文献报道长时间的MIRI,ERK1/2以介导损伤细胞为主,而在早期的损伤中,ERK参与细胞的内源性保护,ERK信号是RAGE下游通路的部分组成成分[21]。在哮喘疾病模型中,已经证实HMGB1可以通过激活RAGE及其下游信号ERK1/2起到损伤呼吸道上皮屏障功能的作用。而本实验证实,FZM1+I/R组的P-ERK水平明显比I/R组高(总的ERK在各组之间无明显差异),可推断出阻断HMGB1与RAGE的结合会进一步激活其下游信号成分ERK1/2,从而起到保护MIRI的作用。不少文献报道药物干预间接激活ERK1/2后,可有效减轻心肌梗死[22],由此根据本实验结果可解释为:小鼠MIRI给予FZM1干预后,先抑制了HMGB1与RAGE信号传导轴,后激活二者的下游信号分子ERK1/2。
综上所述,药物FPS-ZM1可以通过阻断HMGB1-RAGE 信号轴,激活其下游信号是MAPK/ERK成分,减轻由MIRI导致的心肌坏死、水肿及炎症的程度,并且能改善心脏收缩功能。以上结果表明,RAGE很可能成为临床上预防MIRI的一个潜在治疗靶点。
[]
[1] Aleshin A, Ananthakrishnan R, Li Q,et al.RAGE modulates myocardial injury consequent to LAD infarction via impact on JNK and STAT signaling in a mu-rine mode[J].AM J Physiol Heart Circ Physiol,2008,294(4):1823-1832.
[2] Hao M, Zhu S, Hu L,et al.Myocardial ischemic postconditioning promotes au-tophagy against ischemia reperfusion injury via the activation of the nNOS/AMPK/mTOR pathway[J].Int J Mol Sci, 2017,18(3): pii: E614.
[3] Jennings RB.Historical perspective on the pathology of myocardial ischemia/reperfusion injury[J].Circ Res, 2013,113(4):428-438.
[4] Zhang J, Zhang J, Yu P,et al.Remote ischaemic preconditioning and sevoflu-rane postconditioning synergistically protect rats from myocardial injury induced by ischemia and reperfusion partly via inhibition TLR4/MyD88/NF-κB signaling pathway[J].Cell Physiol Biochem, 2017,41(1):22-32.
[5] Zhu J, Yao K, Wang Q,et al.Ischemic postconditioning-regulated miR-499 protects the rat heart against ischemia/reperfusion injury by inhibiting apoptosis through PDCD4[J].Cell Physiol Biochem,2016,39(6):2364-2380.
[6] Tekabe Y, Luma J, Li Q,et al.Imaging of receptors for advanced glycation end products in experimental myocardial ischemiaand reperfusion injury[J]. JACC Cardiovasc Imaging, 2012,5(1):59-67.
[7] Deane R, Singh I, Sagare AP,et al.A multimodal RAGE-specific inhibitor reduces amyloid β-mediated brain disorder in a mouse model of Alzheimer disease [J].J Clin Invest,2012,122(4):1377-1392.
[8] Hong Y, Shen C, Yin Q,et al.Effects of RAGE-specific inhibitor FPS-ZM1 on amyloid-β metabolism and ages-induced inflammation and oxidative stress in rat hippocampus[J].Neurochem Res,2016,41(5):1192-1199.
[9] Bucciarelli LG, Kaneko M, Ananthakrishnan R,et al.Receptor for advanced-glycation end products: key modulator of myocardial ischemic injury[J].Circulation,2006,113(9):1226-1234.
[10] Jordan JE, Zhao ZQ, Vinten-Johansen J.The role of neutrophils in myocardial ischemia-reperfusion injury[J].Cardiovasc Res,1999,43(4):860-878.
[11] Hu G, Zhang Y, Jiang H,et al.Exendin-4 attenuates myocardial ischemia and reperfusion injury by inhibiting high mobility group box 1 protein expression[J].Cardiol J,2013,20(6):600-604.
[12] Andersson U, Wang H, Palmblad K, et al.High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes[J].J Exp Med, 2000,192(4):565-570.
[13] Herzog C, Lorenz A, Gillmann HJ,et al.Thrombomodulin’s lectin-like domain reduces myocardial damage by interfering with HMGB1-mediated TLR2 signalling[J].Cardiovasc Res,2014,101(3):400-410.
[14] Ding HS, Yang J, Gong FL,et al.High mobility group box 1 mediates neutrophil recruitment in myocardial ischemia—reperfusion injury through toll like receptor 4-related pathway[J].Gene,2012,509(1):149-153.
[15] Nogueira-Machado JA, de Oliveira Volpe CM.HMGB-1 as a target for inflammation controlling[J].Recent Pat Endocr Metab Immune Drug Discov,2012,6(3):201-209.
[16] Wu X, Gu W, Lu H, et al.Soluble receptor for advanced glycation end product ameliorates chronic intermittent hypoxia induced renal injury, inflammation and apoptosis via P38/JNK signaling pathways[J].Oxid Med Cell Longev, 2016:1015390. Epub 2016 Sep 5.
[17] Li JZ,Xie MQ,Mo D,et al.Picroside Ⅱ protects myocardium from ischemia/reperfusion-induced injury through inhibition of the inflammatory response [J].Exp Ther Med,2016,12(6):3507-3514.
[18] Li JZ, Wu JH, Yu SY,et al.Inhibitory effects of paeoniflorin on lysophosphatidylcholine induced inflammatory factor production in human umbilical vein endothelial cells[J].Int J Mol Med, 2013,31(2):493-497.
[19] Villarreal A, Aviles Reyes RX, Angelo MF,et al.S100B alters neuronal survival and dendrite extension via RAGE-mediated NF-κB signaling[J].J Neurochem,2011,117(2):321-32.
[20] Shimada K, Nakamura MIshida E.Roles of p38-and c-jun NH2-terminal kinase mediated pathways in 2-methoxyestradiol-induced p53 induction and apoptosis [J].Carcinogenesis, 2003,24(6):1067-1075.
[21] Muth IE, Zschüntzsch J, Kleinschnitz K,et al.HMGB1 and RAGE in skeletal muscle inflammation: Implications for protein accumulation in inclusion body myositis [J].Exp Neurol,2015,271:189-197.
[22] Bourke L, McCormick J, Taylor V, et al.Hydroxychloroquine protects against cardiac ischaemia/reperfusion injury in vivo via ehancement of ERK1/2 phosphorylation[J].PLoS One, 2015,10(12):e0143771.
猜你喜欢
昆明医科大学学报(2022年2期)2022-03-29
中国生殖健康(2020年7期)2020-12-10
工业催化(2020年9期)2020-11-13
无机化学学报(2020年7期)2020-07-20
中国临床医学影像杂志(2019年1期)2019-04-25
无机化学学报(2018年8期)2018-08-01
哈尔滨医药(2016年3期)2016-12-01
中西医结合心脑血管病杂志(2016年20期)2016-03-01
医学研究杂志(2015年2期)2015-06-10
郑州大学学报(医学版)(2015年1期)2015-02-27
