
PPARγ对1型糖尿病心肌病小鼠心肌纤维化的抑制作用及机制
2018-05-28 08:33赵凯潘磊李晓增苗博乔香玲张青青李欣
山东医药 2018年17期
赵凯,潘磊,李晓增,苗博,乔香玲,张青青,李欣
(邢台市第三医院,河北邢台054000)
糖尿病心肌病(DCM)是一种不同于冠心病和高血压引起的心肌病,能够引起微小但持续性的心脏损伤并最终发展为心力衰竭[1,2]。心肌纤维化是其主要的病理学特征。转化生长因子-β1/细胞外信号调节激酶(TGF-β1/ERK)信号通路在心脏疾病中扮演至关重要的角色,而且与胶原生成关系密切[3]。TGF-β1可以维持组织稳态,当其增多时会导致纤维母细胞和平滑肌细胞增殖、炎症和纤维化。有报道指出,过氧化物酶体增殖物激活受体γ(PPARγ)可以抑制肝脏和肺脏的纤维化水平[4],而其在心脏组织中的作用研究较少。2016年6月~2017年12月,本研究拟探讨PPARγ对1型糖尿病心肌病小鼠心肌纤维化的抑制作用及机制。
1 材料与方法
1.1 动物分组及处理 选取12周龄的C57BL/6小鼠60只,平均体质量为25 g,随机分为空白对照组、糖尿病对照组、吡格列酮组、PPARγ抑制剂组、ERK抑制剂组,每组12只。除空白对照组外其余4组每天按照50 mg/kg的剂量腹腔注射链脲霉素,连续5 d,建立1型糖尿病模型。小鼠尾尖取血测量血糖,超过16.6 mmol/L即为造模成功。吡格列酮组每天按20 mg/kg的剂量口服PPARγ的激活剂吡格列酮,连续12周;PPARγ抑制剂组每天按1 mg/kg的剂量腹腔注射PPARγ的抑制剂GW9662,连续12周;ERK抑制剂组每天按1 mg/kg的剂量腹腔注射ERK的抑制剂PD098059,连续12周。
1.2 左室射血分数(LVEF)及早期二尖瓣流入速度比值(E/A)检测 12周后采用异氟烷气体麻醉小鼠,采取5个单位的气流量进行麻醉,2个单位的气流量维持麻醉。采用高分辨自动记录仪测量各组小鼠连续三个心动周期的LVEF和E/A。
1.3 心肌纤维化程度检测 12周后处死小鼠,开胸后无菌分离心脏,采用Masson三色染色,通过检测心肌组织胶原含量,反映各组小鼠心肌纤维化程度。小鼠心脏取下后浸泡在4%的多聚甲醛中24 h,然后流水冲洗过夜。取心脏纵轴中间部分进行石蜡包埋。用20 μm的厚度修片,然后切成5 μm的厚度常温进行保存。取石蜡切片,梯度酒精脱蜡后进行苏木素染色、分化、Masson复合染色、磷钨酸处理、苯胺蓝复染、冰醋酸处理、95%酒精多次脱水、100%酒精脱水、二甲苯透明处理、中性树胶封片等过程,最后用奥林巴斯显微镜拍片。用Image Pro plus软件进行统计分析,用阳性区域面积占总面积的百分比表示心肌组织胶原含量。
1.4 心肌组织TGF-β1表达检测 采用免疫组织化学染色。取心肌组织石蜡切片,梯度乙醇脱蜡后用柠檬酸盐缓冲液进行抗原修复,晾至常温,3%H2O2阻断内源性过氧化物酶活性,然后滴加PBS溶解的TGF-β1抗体,4 ℃孵育过夜。第2天用二抗试剂盒孵育二抗(购自中山金桥公司),然后进行DAB显色,苏木素染核、梯度乙醇脱水透明、封片,最后用显微镜拍照,用Image Pro plus软件进行统计分析,用阳性区域面积占总面积的百分比表示心肌组织TGF-β1表达量。
1.5 心肌细胞培养 分别将各组心肌组织于DMEM培养基中剪碎,在37 ℃用2型胶原酶消化3 h,离心后收集细胞,将其接种于含10%胎牛血清、100 U/mL青霉素和100 g/mL链霉素的培养基中,置于37 ℃含5% CO2的培养箱培养2 h,然后换液弃掉没有贴壁的心肌细胞以及血细胞等杂质。各组细胞培养24 h后收集细胞进行下一步实验。
1.6 心肌细胞PPARγ、ERK、磷酸化ERK(P-ERK)、TGF-β1蛋白表达检测 采用Western blotting法。将不同组处理后的细胞冲洗后提取总蛋白,经过上样、电泳、转膜、封闭后与相应一抗结合,过夜后与二抗结合,然后化学发光检测。所采用的一抗为抗PPARγ抗体、抗总ERK和P-ERK抗体(均购自Cell Signaling Technology公司)以及抗TGF-β1抗体(购自abcam公司),所用二抗为中山金桥公司的抗兔二抗,化学发光液购自Merck公司。图片曝光保存使用GE公司全自动曝光机,灰度值使用photoshop软件进行分析。
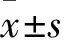
2 结果
2.1 各组小鼠LVEF及E/A比较 诱发糖尿病的糖尿病对照组、吡格列酮组、PPARγ抑制剂组、ERK抑制剂组小鼠LVEF及E/A均低于空白对照组(P均<0.05);吡格列酮组及ERK抑制剂组的LVEF及E/A均高于糖尿病对照组(P均<0.05);PPARγ抑制剂组的LVEF及E/A均低于糖尿病对照组(P均<0.05)。见表1。
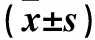
表1 各组小鼠LVEF及E/A比较
2.2 各组心肌纤维化程度比较 空白对照组心肌组织胶原含量为2.47%±0.72%,糖尿病对照组为14.13%±1.72%,吡格列酮组为7.15%±1.38%,PPARγ抑制剂组为17.85%±0.53%,ERK抑制剂组为6.21%±0.44%。诱发糖尿病的糖尿病对照组、吡格列酮组、PPARγ抑制剂组、ERK抑制剂组小鼠心肌组织胶原含量均高于空白对照组(P均<0.05);吡格列酮组及ERK抑制剂组小鼠心肌组织胶原含量均低于糖尿病对照组(P均<0.05);PPARγ抑制剂组小鼠心肌组织胶原含量高于糖尿病对照组(P<0.05)。
2.3 各组心肌组织TGF-β1表达比较 空白对照组心肌组织TGF-β1表达为5.13%±0.71%,糖尿病对照组为17.73%±1.65%,吡格列酮组为8.25%±1.39%,PPARγ抑制剂组为18.45%±1.34%,ERK抑制剂组为8.83%±2.03%。诱发糖尿病的糖尿病对照组、吡格列酮组、PPARγ抑制剂组、ERK抑制剂组小鼠TGF-β1表达均高于空白对照组(P均<0.05);吡格列酮组及ERK抑制剂组TGF-β1的表达低于糖尿病对照组(P均<0.05);PPARγ抑制剂组高于糖尿病对照组(P<0.05)。
2.4 各组心肌细胞PPARγ、ERK、P-ERK、TGF-β1蛋白表达比较 空白对照组、糖尿病对照组以及ERK抑制剂组PPARγ蛋白表达量无统计学差异(P均>0.05);吡格列酮组PPARγ蛋白表达高于糖尿病对照组, PPARγ抑制剂组低于糖尿病对照组,差异均具有统计学意义(P均<0.05)。ERK蛋白表达在各组间无统计学差异(P>0.05)。空白对照组、吡格列酮组、ERK抑制剂组P-ERK、TGF-β1蛋白表达均低于糖尿病对照组,PPARγ抑制剂组高于糖尿病对照组差异均有统计学意义(P均<0.05)。见表2。
表2 各组PPARγ、ERK、P-ERK、TGF-β1蛋白表达比较
3 讨论
DCM的主要特征是心室顺应性下降,舒张功能降低,通过组织学检测可以发现心肌细胞肥大、基质纤维化增加、心肌细胞凋亡[5]。高血糖和胰岛素抵抗引起的代谢紊乱和细胞外基质聚集导致了DCM的进展[6]。持续的高糖刺激可以引起明确的心室重构,减少音猬因子诱导的单核细胞趋化性[7],并且减少基质金属蛋白酶的活性[8],而基质金属蛋白酶具有降解细胞外基质的作用,从而导致细胞外基质的增多,最终引起器官的纤维化[9]。
PPARγ是一种细胞核激素受体,同时也是一种转录因子[10],某些针对肝脏和肺脏组织的研究发现其对胶原的异常增多有一定的抑制作用,但是PPARγ对于心脏纤维化的作用鲜有报道[11]。有人用血管紧张素Ⅱ持续泵入的方法构建了心肌纤维化的模型,发现PPARγ可以通过减少巨噬细胞浸润减轻纤维化[12],而PPARγ对于DCM引起的纤维化尚无报道。我们的实验正是立足于这一点,通过PPARγ激动剂和抑制剂的使用检验其对于DCM纤维化的影响。除PPARγ外,TGF-β1/ERK信号通路与胶原的生成十分密切并且在心脏疾病中扮演至关重要的角色。因此,我们提出激活PPARγ抑制DCM的纤维化其可能的机制是否与抑制TGF-β1/ERK有关。
我们的实验采用了注射链脲霉素的方式构建小鼠DCM的模型。小鼠成模后进行不同种类的药物干预,检测激活PPARγ对于糖尿病心肌纤维化的影响以及可能的分子机制。我们首先检测了小鼠的血糖水平,证明糖尿病模型成功,然后用超声的方法验证了小鼠的心脏功能,诱发糖尿病的4组小鼠LVEF和E/A均低于空白对照组,表明糖尿病小鼠的心脏收缩、舒张功能都有所下降,然而经过吡格列酮处理后心功能有所好转,并且这种好转可以被ERK的抑制剂PD098059所模拟,这就表明PPARγ的激活可能与ERK的抑制有关。我们用Western blotting法检测了PPARγ和ERK的表达,结果证明吡格列酮可以明显提高PPARγ的水平,而GW9662可以抑制PPARγ的水平;PD098059可以抑制而P-ERK,表明我们选取的PPARγ激活剂和抑制剂都是有效的
胶原含量的变化可以直观的反映纤维化,Masson染色结果表明吡格列酮可以减轻糖尿病小鼠的心脏纤维化,表明PPARγ激活可以直接抑制糖尿病心肌纤维化,而ERK抑制剂PD098059可以模拟吡格列酮的作用,表明激活PPARγ可能通过抑制ERK发挥作用。组织切片和Western blotting对于TGF-β1表达的检测发现DCM可以引起TGF-β1的异常增多,表明TGF-β1参与了这一过程。TGF-β1被认为是调节细胞外基质合成最重要的分子[13],其表达异常增多往往伴随组织的纤维化,例如肝脏纤维化、肺脏纤维化都能检测到TGF-β1的过度激活。有研究发现TGF-β1与上皮细胞向间质细胞转化(EMT)还有密切关系[14]。EMT可以分为三型,Ⅰ型主要发生在胚胎时期;Ⅱ型发生在病理状态下某些类型的细胞丢失,并且由间质细胞取代,导致组织器官的纤维化;Ⅲ型主要指的是肿瘤细胞的浸润迁移[15]。因此,DCM时仍然可能存在Ⅱ型EMT。在心脏组织,胶原主要由肌成纤维细胞分泌,而肌成纤维细胞可以由成纤维细胞和内皮细胞通过EMT转化而来。在这个意义上说,吡格列酮抑制了TGF-β1的表达也就抑制了心脏组织中的EMT,大大减少了心脏组织病理状态下的纤维化。
综上所述,我们的实验采用了PPARγ的激活剂和抑制剂以及ERK的抑制剂,证明激活PPARγ可以减轻DCM的心肌纤维化,而抑制PPARγ则加重心肌纤维化,同时,抑制ERK亦可以减轻纤维化,表明激活PPARγ减轻DCM心肌纤维化的主要机制是通过抑制TGF-β1/ERK发挥作用,为临床治疗DCM指明了思路。
参考文献:
[1] Bugger H, Bode C. The vulnerable myocardium[J].Hamostaseologie, 2015,35(1):17-24.
[2] Trachanas K, Sideris S, Aggeli C, et al. Diabetic cardiomyopathy: from pathophysiology to treatment[J]. Hellenic J Cardiol,2014,55(5):411-421.
[3] Munjal C, Jegga AG, Opoka AM, et al. Inhibition of MAPK-Erk pathway in vivo attenuates aortic valve disease processes in Emilin1-deficient mouse model[J]. Physiol Rep, 2017,5(5):e13152.
[4] Mangoni M, Sottili M, Gerini C, et al. A PPAR-gamma agonist attenuates pulmonary injury induced by irradiation in a murine model[J]. Lung Cancer, 2015,90(3):405-409.
[5] Ti Y, Xie GL, Wang ZH, et al. TRB3 gene silencing alleviates diabetic cardiomyopathy in a type 2 diabetic rat model[J]. Diabetes, 2011,60(11):2963-2974.
[6] Falc o-Pires I, Leite-Moreira AF. Diabetic cardiomyopathy: understanding the molecular and cellular basis to progress in diagnosis and treatment[J]. Heart FailRev, 2012,17(3):325-344.
[7] Dunaeva M, Voo S, van Oosterhoud C, et al.Sonic hedgehog is a potent chemoattractant for human monocytes:diabetes mellitus inhibits Sonic hedgehog-induced monocytechemotaxis[J]. Basic Res Cardiol, 2010,105(1):61-71.
[8] Van LS, Seeland U, Riad A, et al. Reduced MMP-2 activity contributes tocardiac fibrosis in experimental diabetic cardiomyopathy[J]. BasicRes Cardiol,2008,103(4):319-327.
[9] Asbun J, Villarreal FJ. The pathogenesis of myocardialfibrosis in the setting of diabetic cardiomyopathy[J]. J Am CollCardiol, 2006,47(3):693-700.
[10] Galli A, Crabb DW, Ceni E, et al. Antidiabetic thiazolidinediones inhibit collagen synthesis and hepatic stellate cell activation in vivo and in vitro[J]. Gastroenterology, 2002,122(7):1924-1940.
[11] Korolczuk A, Maciejewski M, Smolen A, et al. The role of peroxisome-proliferator-activating receptor gamma agonists: rosiglitazone and 15-deoxy-delta12, 14-prostaglandin J2 in chronic experimental cyclosporine A-induced nephrotoxicity[J]. J Physiol Pharmacol, 2014,65(6):867-876.
[12] Caglayan E, Stauber B, Collins AR, et al. Differential roles of cardiomyocyte and macrophage peroxisome proliferator-activated receptor gamma in cardiac fibrosis[J]. Diabetes, 2008,57(9):2470-2479.
[13] Meschiari CA, Ero OK, Pan H, et al. The impact of aging on cardiac extracellular matrix[J]. Geroscience, 2017,39(1):7-18.
[14]Goumans MJ, Mummery C. Functional analysis of the TGFβ receptor/Smad pathway through gene ablation in mice[J]. Int J Dev Biol, 2000,44(3):253-265.
[15] Sun B, Zhang D, Zhao N, et al. Epithelial-to-endothelial transition and cancer stem cells: two cornerstones of vasculogenic mimicry in malignant tumors[J]. Oncotarget, 2017,8(18):30502-30510.
猜你喜欢
渔业研究(2018年5期)2018-01-17
中国卫生标准管理(2015年15期)2016-01-15
中国继续医学教育(2015年6期)2016-01-07
天津医科大学学报(2015年3期)2015-06-05
天津医科大学学报(2015年3期)2015-06-05
中国医疗美容(2015年4期)2015-04-27
郑州大学学报(医学版)(2015年2期)2015-02-27
现代检验医学杂志(2015年2期)2015-02-06
中国药业(2014年17期)2014-05-26
中国合理用药探索(2011年9期)2011-03-20
