
PCSK9表达下调对人脐静脉内皮细胞内质网应激-自噬途径的影响
2018-05-28 08:33龚慧琴赵红刘录山曹建刚姚平波文红艳
山东医药 2018年17期
龚慧琴,赵红,刘录山,曹建刚,姚平波,文红艳
(1南华大学附属南华医院,湖南衡阳421002;2南华大学护理学院;3南华大学心血管病研究所·动脉硬化学湖南省重点实验室)
目前研究发现,细胞自噬、炎症介质、内质网应激等信号通路能够调控动脉粥样硬化[1~3]。自噬在动脉粥样硬化形成过程中扮演着重要的角色,Yamagata等[4]研究证实肿瘤坏死因子信号通路可调控血管内皮细胞自噬促血管细胞黏附分子-1 (VCAM-1)、自噬微管相关蛋白轻链3A(LC3A)及自噬微管相关蛋白轻链3B(LC3B)的表达。如何有效调控内质网应激激活血管内皮细胞自噬、拮抗动脉粥样硬化,目前仍然是其靶向药物研究的热点。本课题组前期研究发现,在氧化型低密度脂蛋白(OX-LDL)诱导THP-1巨噬细胞的细胞模型中,前蛋白转化酶枯草溶菌素9(PCSK9)可介导炎症信号通路调控动脉粥样硬化[5]。近年来研究发现,OX-LDL诱导人脐静脉内皮细胞(HUVEC)可介导内质网应激,过度内质网应激可诱导细胞自噬,介导动脉粥样硬化的病理生理过程[3]。因此,2017年1月~2017年12月本研究拟探讨PCSK9表达下调对HUVEC内受内质网应激-自噬途径的影响。
1 材料与方法
1.1 主要试剂与药品 DMEM培养液(Sigma-Aldrich美国公司产品);胎牛血清(20%FBS,Gibco 美国公司产品);PCSK9引物、PCSK9模拟物、PCSK9抑制物及PCSK9阴性对照物均由美国Sigma公司合成;GRP78、CHOP、LC3B、Beclin-1、VCAM-1及ICAM-1等兔多克隆抗体(Santa Cruz, USA)。
1.2 HUVEC培养及分组 HUVEC(购自中国典型培养物保藏中心),10% FBS的DMEM高糖培养基,放置在37 ℃ (5%CO2)细胞培养箱中培养,24 h给细胞换培养基1次,待细胞密度达到70%~80%时进行细胞传代培养,将培养瓶中的培养液吸出, 加PBS液到瓶中清洗细胞表面2次,弃掉清洗液,加入0.25%胰蛋白酶1.0 mL, 将消化液均匀覆盖细胞表面,消化时间为2 min[5]。进一步观察下调PCSK9是否参与诱导内质网应激介导的HUVEC自噬,将其分为对照组:加 5 μL Hieff TransTM转染试剂;PCSK9模拟组:加5 μL Hieff TransTM转染试剂和100 nmol/L PCSK9模拟物(慢病毒介导的基因转染干扰增强PCSK9);PCSK9抑制组:加5 μL Hieff TransTM转染试剂和100 nmol/L PCSK9抑制物(RNA干扰沉默PCSK9)。转染方法详细见试剂盒(美国Sigma公司产品)说明书操作。
1.3 细胞内GRP78、CHOP、LC3B、Beclin-1、VCAM-1及ICAM-1蛋白表达检测 采用Western blotting法。募集各组细胞后将其PBS洗涤3次,加入裂解液裂解悬浮细胞,然后置于冰上0.5 h后,将其在4 ℃下离心(10 000 r/min,10 min),提取上清液,用BCA法测定蛋白质浓度,随后电泳,采用硝酸纤维素电转膜;5%脱脂牛奶封闭1 h后,加入一抗(GRP78、CHOP、LC3B、Beclin-1、VCAM-1及ICAM-1),4 ℃12 h, TBST洗膜20 min,加入辣根过氧化物酶标记的二抗,室温孵育2 h,TBST洗膜10 min,然后用显色(化学发光法),采集图像(UVP 型凝胶图像分析系统),分析条带积分吸光度(Imag J软件)。以对照组为参照计算蛋白的表达量。
1.4 细胞内GRP78、CHOP、LC3B、Beclin-1、VCAM-1及ICAM-1 mRNA表达检测 采用qRT-PCR法。提取各组细胞RNA(TRIzol法),逆转录得到cDNA,然后行PCR循环取RT-PCR产物电泳(1.2%琼脂糖凝胶),GRP78、CHOP、LC3B、Beclin-1、VCAM-1及ICAM-1等加样后溴化乙锭染色,采用UVP型凝胶图像分析电泳条带并系统做积分吸光度测定和分析。目的基因的表达量通过标化内参后分析。基因引物序列由上海生物工程有限公司合成。
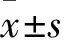
2 结果
2.1 各组HUVEC GRP78、CHOP、LC3B、Beclin-1、VCAM-1及ICAM-1蛋白表达比较 与对照组比较,PCSK9抑制组GRP78,CHOP、LC3B、Beclin-1蛋白表达均升高,VCAM-1、ICAM-1蛋白表达均降低,而PCSK9模拟组结果与之相反(P均<0.05)。见表1。

表1 各组HUVEC细胞GRP78、CHOP、LC3B、Beclin-1、VCAM-1及ICAM-1蛋白表达比较(±s)
注:与对照组比较,aP<0.05。
2.2 各组HUVEC GRP78、CHOP、LC3B、Beclin-1、VCAM-1及ICAM-1 mRNA表达比较 与对照组比较,PCSK9抑制组GRP78,CHOP、LC3B、Beclin-1 mRNA表达均升高,VCAM-1、ICAM-1 mRNA表达均降低,而PCSK9模拟组结果与之相反(P均<0.05)。见表2。
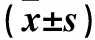
表2 各组HUVEC GRP78、CHOP、LC3B、Beclin-1、VCAM-1及ICAM-1 mRNA表达比较
注:与对照组比较,aP<0.05。
3 讨论
丝氨酸激酶PCSK9可与介导LDL受体(LDLR)上的表皮生长因子A区域结合并促进LDLR降解,因此下调PCSK9可有效阻止PCSK9与LDLR结合,从而发挥降胆固醇作用;Xu等研究证实:下调PCSK9的表达可介导内质网应激及线粒体信号通路激活 caspase-3的表达从而起到抑制肺癌的作用[6]。近年来证据表明:内质网应激-自噬信号通路介导凋亡相关酶的去泛素化修饰或磷酸化修饰来调控内质网应激的稳态,起到改善糖尿病大鼠心肌纤维化的作用[7]。因此,本研究进一步探讨PCSK9是否调控内质网应激反应,激动细胞自噬抗动脉粥样硬化;是否与PCSK9的调控内质网应激-自噬信号通路密切相关将是本研究的新视角。
内质网是重要的细胞器之一,许多疾病的发生发展都与内质网的功能稳态息息相关。大量研究表明:缺血/再灌注损伤、炎症介质爆炸性释放、氧化应激、自噬、Ca2+通道等内环境失衡可介导内质网功能紊乱诱导内质网应激[8]。内质网应激可启动内质网未折叠蛋白反应(标志物如GRP78、CHOP)和诱导自噬信号通路[9,10]。研究也表明:Beclin-1与LC3B与动脉粥样硬化斑块的稳定性密切相关,适度自噬可提升斑块内细胞对氧化应激的抵抗力,促细胞恢复及细胞保护性作用[10];研究也证实:适度的内质网应激可通过细胞自噬或去泛素化修饰未折叠或错误折叠蛋白,恢复机体内环境稳态,维持细胞的正常生命活动[9~11]。因此,内质网应激-自噬途径可能是参与动脉粥样硬化发生发展过程中的关键的病理生理机制之一。本实验结果证实:GRP78、CHOP、Beclin-1与LC3B表达的演变趋势呈现了内质网应激-自噬信号通路的细胞自噬活性。
研究发现:下调PCSK9可在转录后水平特异性靶向甄别靶点基因LDLR,以不同的修饰方式抑制或加速降解靶点基因而发挥抗动脉粥样硬化的作用[5]。且研究发现:过度内质网应激可介导粘附分子(VCAM-1)及细胞间粘附分子(ICAM-1)的表达促动脉粥样硬化[8]。本研究结果提示:下调PCSK9的表达可调控内质网应激与自噬之间的"杠杆原理"从而下调VCAM-1、ICAM-1的表达起到抗动脉粥样硬化作用,与本课题前期研究一致。研究表明PCSK9可调控ERK/INK/MAPK/mTOR信号通路介导凋亡因子半胱天冬酶的激活,也可调控内质网应激-自噬信号转导机制加重脏器的损伤,故调控PCSK9的表达可减轻上述病理生理过程[3,4,6,12,13]。鉴于PCSK9广泛参与生物学效应的及调控心脏的生理病理过程的事实。因此,我们推测PCSK9可能参与了内质网应激介导HUVEC细胞自噬的调控。
为此,我们对HUVEC细胞的PCSK9基因的表达予以调控。研究表明:PCSK9低表达的抑制组GRP78、CHOP的表达均明显上调,提示下调PCSK9增加内质网应激介导的内质网应激性细胞自噬,低表达PCSK9调控内质网应激-自噬之间的稳态从而抗动脉粥样硬化。而模拟组GRP78、CHOP的表达均明显下调,提示上调PCSK9可减少内质网应激介导的细胞自噬,高表达PCSK9抑制细胞自噬调控内质网应激的稳态从而促动脉粥样硬化。上述研究结果提示上调PCSK9介导内质网应激-自噬途径参与了动脉粥样硬化的病理生理过程。本研究从PCSK9转录后调控这一新视角揭示其精准调控内质网应激-自噬的可能信号转导机制,这将为临床上的靶向药物研发提供新的证据。但PCSK9如何调控的细胞自噬,进而调节内质网应激-自噬的机制及信号通路尚须更进一步深入的研究。
综上所述,本研究表明下调PCSK9介导内质网应激-自噬途径可调控动脉粥样硬化,其机制涉及细胞自噬与内质网的应激之间的稳态起到保护血管内皮的作用。此外,本研究还证实靶向调控PCSK9基因平衡上述稳态的作用从而抑制动脉粥样硬化。
参考文献:
[1] Viona AC, Kheloufia M, Hammoutene A, et al. Autophagy is required for endothelial cell alignment and atheroprotection under physiological blood flow[J]. Proc Natl Acad Sci U S A, 2017,114(41):8675-8684.
[2] Hofmann A, Brunssen C, Morawietz H. Contribution of lectin-like oxidized low-density lipoprotein receptor-1 and LOX-1 modulating compounds to vascular diseases [J]. Vascul Pharmacol, 2017, 1537-1891(17):30171-30174.
[3] Kheloufi M, Vion AC, Hammoutene A, et al. Endothelial autophagic flux hampers atherosclerotic lesion development[J]. Autophagy, 2018, 14(1):173-175.
[4] Yamagata K, Xie Y, Suzuki S,et al. Epigallocatechin-3-gallate inhibits VCAM-1 expression and apoptosis induction associated with LC3 expressions in TNFα-stimulated human endothelial cells[J]. Phytomedicine, 2015,22(4):431-437.
[5]刘录山,程艳丽,谢闵等. LDL、ox-LDL对THP-1巨噬细胞PCSK9、LDLR表达的影响研究[J].生物化学与生物物理进展,2008,35(5):540-547.
[6] Xu X, Cui Y, Cao L,et al.PCSK9 regulates apoptosis in human lung adenocarcinoma A549 cells via endoplasmic reticulum stress and mitochondrial signaling pathways[J]. Exp Ther Med, 2017,13(5):1993-1999.
[7] Liang B , Xiao T , Long J , et al. Hydrogen sulfide alleviates myocardial fibrosis in mice with alcoholic cardiomyopathy by downregulating autophagy[J]. Int J Mol Med, 2017,40(6):1781-1791.
[8] Shapiro MD, Fazio S. PCSK9 and Atherosclerosis-Lipids and Beyond[J]. Atheroscler Thromb, 2017, 24(5): 462-472.
[9] Seidah NG. The PCSK9 revolution and the potential of PCSK9-based therapies to reduce LDL-cholesterol [J]. Glob Cardiol Sci Pract, 2017,19(1):1702-1711.
[10] Du Y, Li S, Cui CJ, et al. Leptin decreases the expression of low-density lipoprotein receptor via PCSK9 pathway: linking dyslipidemia with obesity[J]. Transl Med, 2016,14(11): 276-283.
[11] Luan Q, Jin L, Jiang CC, et al. RIPK1 regulates survival of human melanoma cells upon endoplasmic reticulum stress through autophagy[J]. Autophagy, 2015,11(7): 975-994.
[12] Glerup S, Schulz R, Laufs U, et al. Physiological and therapeutic regulation of PCSK9 activity in cardiovascular disease[J].Basic Res Cardiol, 2017,112(3): 32-41.
[13] Ding Z, Liu S, Wang X, et al. Hemodynamic Shear Stress via ROS Modulates PCSK9 Expression in Human Vascular Endothelial and Smooth Muscle Cells and Along the Mouse Aorta[J]. Antioxid Redox Signal, 2015,22(9):760-771.
猜你喜欢
今日农业(2022年14期)2022-09-15
大电机技术(2022年3期)2022-08-06
核科学与工程(2021年4期)2022-01-12
煤气与热力(2021年4期)2021-06-09
现代临床医学(2021年1期)2021-01-26
中华戏曲(2020年1期)2020-02-12
天津医科大学学报(2019年6期)2019-08-13
制造技术与机床(2017年6期)2018-01-19
安徽医科大学学报(2016年12期)2017-01-15
中西医结合心脑血管病杂志(2016年20期)2016-03-01
