
低氧条件下急性肺损伤小鼠肺血管通透性变化*
2018-05-28 06:35张东冬陈魁君翁昌梅王建民
微循环学杂志 2018年2期
张东冬 陈魁君 翁昌梅 王建民
急性肺损伤(Acute Lung Injury,ALI)是各种致病因素作用下发生的一类严重综合症,致死的主要原因是肺泡腔充满蛋白类炎性渗出液,阻碍肺泡气体交换,从而引起动脉血氧不足以及呼吸窘迫,具有并发症多、死亡率高的特点(死亡率为30%-40%)[1,2]。在低氧条件下,ALI死亡率更高,可达到35%-60%[3]。研究[4,5]发现,低氧与炎症有着相互依存的联系,低氧能够诱发并加重组织炎症,促进肺脏等器官释放炎性细胞因子,使其血液水平升高,导致炎性细胞聚集增加,加重肺水肿。此外,缺氧诱导因子(HIF-1α)是机体缺氧反应中关键的转录激活因子,能够激活下游信号通路,产生促炎细胞因子和趋化因子,参与血管生成、细胞凋亡、炎症反应等一系列病理生理过程[6]。但它们与低氧条件下ALI死亡率高的相关机制并未完全阐明,为明确低氧条件下ALI高死亡率的发生机制,本研究采用低压氧仓模拟低氧环境,从肺间质屏障功能以及肺血管通透性方面来探究低氧环境下ALI死亡率的可能机制。
1 材料与方法
1.1 实验动物与分组
雄性昆明白小鼠280只,8-10周龄, 体重30±3g,来源于陆军军医大学第三附属医院实验动物中心(动物许可证号:SYXK(军)2012-0037)。实验前一周小鼠被分为5只一笼饲喂,室温22±2℃,湿度为50%±10%,自由饮水与进食。实验时,小鼠被随机分成四组:常氧+PBS组(CK组,n=35)、常氧+LPS组(NL组,n=60)、低氧+PBS组(HP组,n=35)、低氧+LPS组(HL组,n=150),其中15只用于测定存活率,剩余小鼠用于其它数据采集。
1.2 主要药物、试剂和仪器
内毒素(LPS,L2630-100mg)、 伊文思蓝染料(EVANS blue dye,E2129-10g)购买于美国Sigma-Aldrich公司(St. Louis, MO);甲酰胺(成都科龙化工试剂);戊巴比妥钠(CAS:57-33-0,Sigma);多聚甲醛(EKKS1699,上海生工);HE染色液(C0105,碧云天);石蜡切片机(莱卡-RM2015);光学显微镜(日本OLYMPUS公司BX75);电子称(赛多利斯-BSA124S);低压氧仓(贵州风雷氧仓有限公司-DYC型)。
1.3 ALI模型制备及各组动物处理
NL组与HL组小鼠按照Zhang等[7]方法,通过腹腔注射LPS(8mg/kg,注射前LPS用生理盐水稀释成2.5μg/μl)的方式诱导ALI模型,CK组和HP组腹腔注射同体积PBS,本文所有造模小鼠全部造模成功。LPS作用后2h,将HP组与HL组小鼠转移至低压氧仓中饲喂,模拟6 000米高原低压低氧环境,CK组与NL组饲喂在动物中心。各组小鼠分别在第1、3、7天通过腹腔注射戊巴比妥钠(1%、0.3ml)的麻醉取材。
1.4 观察指标和方法
1.4.1小鼠肺含水量检测:在造模后第1、3、7天每组每次取3只小鼠,将小鼠接上述方法麻醉后切开胸腔取下全肺,去除多余组织包膜以及心脏,用纱布擦净表面水分与血渍,并用电子称获得肺湿重(W)。后将湿肺放入80℃烤箱中烘烤48h,以获得干肺重量(D)并记录,计算出小鼠肺水含量(Q),Q=(W-D)/W×100%。
1.4.2肺组织病理学分析及损伤评分:在第1、3、7天每组每次取3只小鼠,将小鼠麻醉开胸腔后经肺循环灌注50ml PBS后,用4%的多聚甲醛进行灌注固定,取下动物左肺放入多聚甲醛缓冲液中固定24h,然后在不同浓度梯度的乙醇溶液中分级脱水,石蜡包埋,制成厚5μm的石蜡切片,常规HE染色后进行组织病理学分析。依据Nishina等[8]提出的肺损伤评分(Lung Injury Score, LIS)进行损伤严重度评估。无明显病变计0分(无损伤),病变范围<25%计1分(轻度),病变范围25%-50%计2分(中度),病变范围50-75%计3分(重度),病变范围>75%计4分(极严重)。每个标本在200倍镜下随机选取6个区域,取均值为该标本的LIS分值。
1.4.3肺血管通透性测定:肺血管通透性采用伊文思蓝染料通过血管的渗透性来评估。在第1、3、7天取材前1h,每组选取3只小鼠,通过尾静脉注射伊文思蓝染料(20mg/kg),1h后麻醉小鼠,开胸腔后经肺循环灌注50ml PBS,清洗残留在肺组织中的血液以及染料,摘除肺组织置于90℃烤箱中烘烤24h,称取100mg肺组织放入EP管中,剪碎后加入2ml甲酰胺,并置于50℃恒温水浴锅中加热,24h后离心提取上清液,在620nm波长下检测吸光度,最终计算出组织上清液中伊文思蓝浓度(μg/g),其值越大,表明肺血管通透性越大[9]。
1.4.4各组小鼠存活率观察:小鼠放入低压氧仓中
开始计时,每隔24h观察并记录小鼠存活情况,持续七天。
1.5 统计学处理

2 结果
2.1 各组小鼠存活率比较
CK组与HP组在7天实验观察中均无死亡发生,存活率100.00%。NL组共死亡5只,存活率为66.67%;HL组共死亡11只,存活率26.67%。NL组存活率显著高于HL组(χ2=5.23,P<0.05),且两组死亡均发生在前3天,随后观察时间内无死亡发生(图1)。

图1 各组小鼠生存率曲线
2.2 小鼠肺水含量比较
比较各组第1、3、7天小鼠肺水含量,第1天,各组肺水含量差异无统计学意义(P>0.05);第3天,各组肺水含量差异有统计学意义(P<0.05),NL组和HL组小鼠肺水含量显著高于CK组(t均>7.87,P<0.01);第7天,各组肺水含量差异有统计学意义(P<0.05)和HP组和NL组肺水含量与CK组无显著差异(t均<1.07,P>0.05),HL组则显著高于CK组(t=4.77,P<0.01)与NL组(t=4.77,P<0.01)。见表1。
2.3 各组小鼠肺血管通透性变化
第1天,各组间伊文思蓝含量均无显著差异(P>0.05);第3天,各组间伊文思蓝含量差异有统计学意义(P<0.05),HL组伊文思蓝含量显著高于CK组(t=-4.10,P<0.05),HL组与NL组之间无显著差异(t=-1.41,P>0.05);第7天,各组伊文思蓝含量差异有统计学意义(P<0.01)。其中,HP组、NL组和HL组均高于CK组,HL组最高,明显高于HP组和NL组(t均>-35.31,P<0.01)。见表2。

表1 各组小鼠肺水含量比较均=3)
注:与CK组比较,1)P<0.01;与NL组比较,2)P<0.01
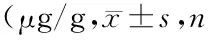
表2 各组小鼠肺组织伊文思蓝含量比较均=3)
注:与CK组比较,1)P<0.05,2)P<0.01;与HP组和NL组比较,3)P<0.01
2.4 各组小鼠肺组织病理学变化
CK组第1、3、7天 (图2A-C)和HP组第1天(图2D)肺脏均未见明显的病理学病变,NL组和HL组第1天小鼠肺泡腔均出现了不同程度损伤(图2G、图2J)。第3天,HP组肺泡间质有轻微增厚(图2E),NL组肺组织水肿,肺泡腔被破坏(图2H),HL组间质明显增厚,并出现轻微肺间质水肿(图2K)。至第7天,HP组肺间质明显增厚,并有轻微水肿现象(图2F),NL组肺泡损伤则逐渐恢复,肺水肿减轻,损伤程度有所下降(图2I),HL组肺间质水肿严重,肺泡腔缩小,肺间质增厚并伴有实化(图2L)。
2.5 各组小鼠肺损伤评分
第1天、第3天和第7天,各组小鼠LIS评分差异均有统计学意义(P<0.05或P<0.01)。第1天,CK组与HP组、NL与HL组LIS评分差异无统计学意义(t<-1.00,P>0.05);但NL组和HL组均较CK组和HP组明显升高(t均>-3.16,P<0.01)。第3天,HP组、NL组、HL组较CK组LIS评分均显著升高(t均>-5.40,P<0.01),且HL组升高更明显,显著高于HP组和NL组(t均>-3.46,P<0.05或P<0.01)。第7天,HP组和NL组较CK组LIS评分均显著升高(t均>-2.74,P<0.05),HL组明显高于其它各组(t均>-3.35,或P<0.05或P<0.01)。见表3。

图2 低氧与常氧环境下肺损伤后病理学比较(HE,×200)
3 讨论
ALI和急性呼吸窘迫综合症(ARDS)是导致患者急性呼吸衰竭和早期死亡的重要原因。ALI常常是由严重感染、创伤等不同病因所致的肺泡毛细血管内皮细胞、肺泡上皮细胞和肺间质的急性弥漫性损害,本质是肺部多种炎性细胞如中性粒细胞(PMN)、肺泡巨噬细胞(Alveolar Macrophage,AM)等所介导的肺部炎症反应及失控的炎症反应所致的肺泡毛细血管损伤[10]。其最基本的病理生理改变就是肺血管内皮细胞及肺泡上皮细胞屏障对液体、蛋白通透性的增加,进而导致肺泡水肿。在低氧条件下,缺氧可诱发高原肺水肿(HAPE), 进一步增加肺泡水肿程度,影响肺的换气功能,加重肺部炎症反应[11]。在临床上常表现为呼吸困难,严重发绀,咳粉红色泡沫痰或白色泡沫痰,肺部有湿啰音等。同时,缺氧能够引起心排出量增加、血流分布改变、肺血管收缩与毛细血管增生,加重肺毛细血管损伤,进而诱发更为严重的ALI[12]。

表3 各组小鼠肺损伤评分均=3)
注:与CK组比较,1)P<0.05,2)P<0.01;与HP组比较,3)P<0.05,4)P<0.01;与NL组比较,5)P<0.05,6)P<0.01
在常氧下,正常的肺组织和血管内皮细胞是分泌和合成血管活性物质的重要场所,血管内皮细胞连续地分布于血管腔内面,它对于维持血管壁完整性、分泌和释放活性物质、维持血管张力等具有重要作用。在本实验中,HL组小鼠肺水含量增加,其发生机制可能是缺氧引起肺血管收缩,肺动脉压增高,肺毛细血管内压增高,周血管收缩,肺血流量增多,血浆、蛋白和红细胞经肺泡-毛细血管壁漏出至间质或肺泡;其次,缺氧时活性氧释放增多、血管内皮生长因子(VEGF)表达上调,以及白细胞介素-1(IL-1)、肿瘤坏死因子-α(TNF-α)等炎性介质释放增多,引起血管内皮细胞通透性增高,液体渗出[11]。HL组小鼠肺血管通透性增加,这可能是由于在LPS、低氧因子及血管活性物质等共同作用下,肺血管内皮细胞功能紊乱甚至出现坏死、脱落等,引起血管内皮细胞受损并致肺泡毛细血管膜损伤进而引起血管壁通透性增加。同时,在低氧条件下,肺血管收缩能够加剧肺毛细管压力升高,升高的血压能够进一步促进渗透,这种血管反应主要发生在肺泡缺氧的条件下。在低气压条件下,液体过滤渗透的压力会比正常的气压下低得多,能够在较低压力下引起组织水肿与渗透。并且,这个过程中可以激活白细胞、巨噬细胞和血小板,间接地引起肺损伤。大量中性粒细胞趋化因子,如TNF-α、白细胞介素-8(IL-8)、脂多糖(LPS)、血小板活化因子(PAF)、纤维蛋白降解产物(FDPs)等作用下,聚集于肺、粘附于肺泡毛细血管内皮,释放氧自由基、蛋白酶和炎性介质等,损伤肺泡上皮细胞及毛细血管内皮细胞。血管内膜的损伤和中性粒细胞及肺组织释放的促凝物质,导致血管内凝血,形成微血栓,后者通过阻断血流进一步引起肺损伤,通过形成纤维蛋白降解产物及释放TXA2等血管活性物质进一步使肺血管通透性增高[12]。因此,HL组小鼠存活率最低。
同时,在实验时将实验鼠直接放入低压氧仓中,这种急性缺氧可能会引起肺血管收缩。研究发现,慢性缺氧在引起肺血管收缩的同时还可引起以管壁增厚、官腔狭窄为特征的肺血管结构改建,导致持续的肺动脉高压。本文也观察到在造模后第7天,HL组小鼠肺泡腔缩小,肺间质增厚并伴实质化。其发生机制主要是长期缺氧可选择性抑制肺动脉Kv通道α亚单位的表达,使外向性K+电流减少,细胞膜去极化,Ca2+内流增加,在引起血管收缩的同时促进平滑肌细胞增殖[13];另一方面,慢性缺氧可引起组织中毛细血管增生,其机制主要在于,缺氧诱导因子-1增多,上调VEGF等基因的表达,进而促进毛细血管增生。另外,肺血管周围肥大细胞密度也会增加,肥大细胞增生的数量和小动脉中层的厚度有较好的相关性。肥大细胞释放某些介质,如组织胺、类胰蛋白酶(Tryptase)等因子都是肥大细胞分泌的一类血管新生因子[11]。在7天的实验过程中,这有可能使肺血管收缩,从而使得血管肥大,肺间质增厚。
综上,低氧环境中缺氧引起的肺血管通透性增加以及慢性缺氧引起的肺间质屏障功能受损可能是低氧条件下ALI死亡率增加的主要原因。
◀
本文第一作者简介:
张东冬(1987-),男,汉族,硕士,助理研究员,主要从事野战外科学的研究
参考文献
1 Pittet JF, Griffiths MJD, Geiser T,et al. TGF-β is a critical mediator of acute lung injury[J]. Clin Invest,2001,107(7):1 537-1 544.
2 Ware LB, Matthay MA. The acute respiratory distress syndrome[J]. N Engl J Med,2000, 342(3):1 334-1 349.
3 Eckle T, Fullbier L, Wehrmann M, et al. Identification of ectonucleotidases CD39 and CD73 in innate protection during acute lung injury[J]. J Immunol, 2007,178(3):8 127-8 137.
4 Hartmann G, Tschop M, Fischer R, et al. High altitude increases circulating interleukin-6, interleukin-1 receptor antagonist and C-reactive protein[J]. Cytokine, 2000,12(1):246-252.
5 Eckle T, Faigle M, Grenz A, et al. A2B adenosine receptor dampens hypoxia-induced vascular leak[J]. Blood, 2008,111(2):2 024-2 035.
6 Eltzschig HK, Eckle T. Ischemia and reperfusion: from mechanism totranslation[J]. Nat Med, 17(1):1 391-1 401.
7 Zhang XL, Li CL, Li J, et al. Protective effects of protocatechuic acid on acute lung injury induced by lipopolysaccharide in mice via p38MAPK and NF-κB signal pathways [J]. International Immunopharmacology, 2015, 26(1):229-236.
8 Nishina K, Mikawa K, Takao Y, et al. ONO-5046, an elastase inhitor, attenuates endotoxin-induced acute lung injury in rabbits[J]. Anesth Analg, 1997,84 (2):1 097-1 103.
9 Koksoy C, Kuzu MA, Kuzu I, et al. Role of tumour necrosis factor in lung injury caused by intestinal ischaemia-reperfusion[J]. Br J Surg, 2001, 88(12):464-480.
10 Atabai K, Matthay MA. The pulmonary physician in critical care. 5:acute lung injury and the acute respiratory dist resssyndrome: definitions and epidemiology[J]. Thorax,2002, 57(3):452-458.
11 王建枝,殷莲华. 病理生理学[M]. 北京:人民卫生出版社,2015:219-230.
12 Pavlicek V, Marti HH, Grad S, et al. Effects of hypobaric hypoxia on vascular endothelial growth factor and the acute phase response in subjects who are susceptible to high-altitude pulmonary oedema[J].Eur J Appl Physiol, 2000,81:497-503.
13 王建枝,殷莲华. 病理生理学[M]. 北京:人民卫生出版社,2015:88-102.
猜你喜欢
中国典型病例大全(2022年7期)2022-04-22
广东医科大学学报(2020年6期)2020-02-06
莫愁·时代人物(2019年3期)2019-03-30
湘潮(2016年5期)2016-10-20
湘潮(上半月)(2016年5期)2016-01-30
畜牧兽医学报(2015年3期)2015-07-05
郑州大学学报(医学版)(2015年2期)2015-02-27
中华皮肤科杂志(2014年3期)2014-12-19
中国民族民间医药·下半月(2014年10期)2014-10-27
中国火炬(2014年8期)2014-07-24
