
氧化修饰多壁碳纳米管固相萃取-超高效液相色谱/串联质谱法同时测定牛奶中7种雌性激素残留
2018-05-07 02:42:52陈万勤徐潇颖赵超群梁晶晶丁宇琦罗金文
分析科学学报 2018年1期
刘 柱, 华 颖, 陈万勤, 徐潇颖, 赵超群,梁晶晶, 丁宇琦, 周 霞, 罗金文*
(1.浙江省食品药品检验研究院,浙江杭州 310052;2.浙江中医药大学,浙江杭州 310053)
雌性激素可调节动物生长模式,诱导奶牛发情和泌乳,提高牛奶产量[1],但牛奶中激素残留会随着日常摄入在人体内蓄积,导致内分泌失调、生殖功能障碍、发育异常等病变[2]。我国农业部176号、193号公告明确规定,禁止饲料和动物饮用水中添加己烯雌酚、雌二醇等雌性激素,农业部235号公告规定己烯雌酚、己烷雌酚等外源性激素物质在动物源性食品中不得检出。因此,建立快速高效、准确灵敏的雌性激素残留检测方法显得十分重要。
目前激素残留的测定方法主要有气相色谱-质谱法(GC-MS)[3]、高效液相色谱法(HPLC)[4]和液相色谱-串联质谱法(LC-MS/MS)[5 - 7]。LC-MS/MS法测定激素时具有高选择性和高灵敏度,且不需要衍生,但牛奶基质复杂,激素的残留量低,因此样品前处理技术成为牛奶中雌性激素检测的关键。多壁碳纳米管(MWCNTs)是一种新型纳米材料,于1991年由日本科学家Iijima首次发现[8],它具有独特的空间结构,比表面积大、理化性质稳定,能通过静电、范德华力和疏水等相互作用与不同性质的化合物相互吸附,自被发现以来就引起了广泛关注[9 - 10],已被成功应用于水体中多环芳烃、农药、磺胺等农兽药污染物的分离富集[11 - 12]。李鱼等[4]使用MWCNTs作为固相萃取柱的填料实现了水样中雌三醇、17α-乙炔基雌二醇及17β-雌二醇等微量激素的富集。然而MWCNTs的疏水性限制了它的应用范围[13]。随着研究的深入,发现MWCNTs两端具有球形富勒烯突起,使用H2SO4和HNO3等强酸处理能产生羟基、羧基等官能团[14],有望实现对甾体类化合物的高效富集。而将MWCNTs改性修饰后再作为固相萃取材料应用于雌性激素的研究尚未见报道。
本文采用氧化性强酸实现对MWCNTs进行表面氧化改性的修饰,再应用固相萃取(SPE)技术实现对牛奶样品中雌性激素的富集、净化,结合超高效液相色谱-串联质谱(UPLC-MS/MS)分析技术建立牛奶中雌性激素的定性、定量检测方法。本研究对于扩展MWCNTs的应用范围,解决牛奶等复杂基质样品的前处理难题具有重要意义。
1 实验部分
1.1 仪器、试剂与材料
AB 5500 QTRAP质谱仪(美国,AB Sciex公司);LC-30AD超高效液相色谱仪(日本,岛津公司);Milli-Q超纯水器(美国,Millipore公司);高速冷冻离心机(美国,Thermo公司);KH-500DV超声仪(昆山禾创超声仪器有限公司);全自动氮吹浓缩仪(美国,Biotage公司)。
雌二醇(98.0%±1.0%)、雌三醇(98.8%±0.5%)、己烯雌酚(99.5%±1.0%)、己烷雌酚(99.8%±0.5%)、己二烯雌酚(96.0%±1.0%)、雌酮(99.5%±0.5%),均购自德国Dr.Ehrenstorfer公司;炔雌醇(≥98%)购自美国SIGMA公司;己烯雌酚-D8(98.9%)、雌酮-D2(99.5%)、己烷雌酚-D4(98.5%)均购自加拿大T.R.C.公司;已二烯雌酚-D2(85.6%)购自北京振翔公司。雌激素标准溶液:准确称取上述雌性激素标准品,分别用甲醇配制成1 000 mg/L的标准储备液,于-18 ℃下避光保存;将上述各单标储备液用甲醇稀释成10 mg/L的混合标准储备液,于-18 ℃下避光保存;使用前用50%甲醇将混合标准储备液稀释成浓度为1 000 μg/L和100 μg/L的混合标准工作液。准确称取上述雌性激素的同位素内标分别用甲醇配制成500 mg/L的同位素内标储备液,于-18 ℃下避光保存;再将上述各同位素内标储备液用甲醇稀释成1 000 μg/L的混合同位素内标工作液,于-18 ℃下保存。甲醇、乙腈、乙酸乙酯、叔丁基甲醚、正己烷,均为色谱纯(Merk公司);乙酸、氨水为色谱纯(Sigma公司);HNO3、H2SO4为优级纯(国药集团化学试剂有限公司);其它试剂均为分析纯。
1.2 MWCNTs固相萃取柱的制备
1.2.1MWCNTs表面修饰处理称取2.0 g MWCNTs粉末,置于500 mL平底烧瓶中,缓慢加入100 mL浓H2SO4,超声分散30 min,加入150 mL浓HNO3,常温超声1 h,于加热反应釜中120 ℃回流1 h,再降至60 ℃回流12 h(过夜),冷却至室温,除去上层酸液,去离子水反复洗至中性,80 ℃干燥,研磨成细粉,即得表面修饰的MWCNTs。
1.2.2装柱及活化取表面修饰MWCNTs粉末150 mg,加入10 mL甲醇,震荡混合使表面修饰MWCNTs粉末分散在甲醇中,快速倒入6 mL固相萃取空柱中,调节甲醇流速为1滴/秒,使MWCNTs均匀填充,放置6 mL固相萃取柱专用柱筛板。使用前依次用5 mL甲醇、5 mL水活化。
1.3 样品前处理
准确称取5.00 g牛奶样品(固体奶粉复原后称取)于50 mL离心管中,加入10 mL乙腈,涡旋混合1 min,超声提取10 min,于4 ℃下15 000 r/min高速离心10 min,取5 mL上清液,用去离子水稀释10倍后全部过固相萃取柱,依次用10 mL水和10 mL 10%甲醇溶液淋洗,抽至近干后,用5 mL的氨水-甲醇(5∶95,V/V) 溶液洗脱,收集洗脱液,重复洗脱一次,合并洗脱液,整个过程控制流速1 mL/min,洗脱液于45 ℃下氮气吹至近干,准确加入1.0 mL初始流动相复溶,过0.22 μm微孔滤膜后,供UPLC-MS/MS分析。
1.4 UPLC-MS/MS分析条件
1.4.1UPLC条件色谱柱:Waters ACQUITY UPLC®BEH C18柱(100×2.1 mm,1.7 μm);柱温:35 ℃;进样体积:5 μL;流速:0.4 mL/min;流动相:0.02%氨水溶液(A相),乙腈(B相);梯度洗脱程序:0~5 min,80%~20%A;5~6 min,20%A;6~6.1 min,20~80%A;6.1~9 min,80%A。
1.4.2MS条件采用多反应监测模式(MRM)检测,负离子模式:喷雾电压4.5 kV,雾化器压力(GS1)345 kPa,辅助气压力(GS2)345 kPa,离子源温度500 ℃,气帘气压力(CUR)275 kPa。优化后的母离子、子离子和锥孔电压、碰撞能量等相关参数见表1。

表1 雌性激素的主要质谱参数和保留时间Table 1 MS parameters and retention time for the analysis of estrogens
*Quantitative ion.
2 结果与讨论
2.1 质谱方法建立
采用蠕动泵将浓度为100 μg/L的雌性激素溶液及同位素内标溶液连续直接进样,在ESI-模式下,确定各目标化合物的母离子和子离子,分别优化毛细管电压、锥孔电压、碰撞能量和碰撞室出口电压等质谱参数,以达到最佳灵敏度。在优化条件下,选择相对丰度较高、响应稳定的离子作为定量离子,优化后的质谱参数详见表1。
2.2 色谱条件的优化

图1 7种雌性激素的总离子流(TIC)色谱图Fig.1 TIC chromatogram of 7 estrogens1.estradiol;2.estriol;3.ethinylestradiol;4.estrone;5.diethylstilbestrol;6.dienestrol;7.hexestrol.
鉴于目标化合物都含-OH或-CO-的类甾体物质,极性中等偏弱,实验选择Water公司ACQUITY UPLC®BEH C18柱(100×2.1 mm,1.7 μm)、Cortecs C18柱(100×2.1 mm,2.7 μm)和HSS T3柱(100×2.1 mm,1.8 μm)三种不同类型的色谱柱进行考察。结果发现ACQUITY UPLC®BEH C18色谱柱对目标化合物的分析效果较好,保留时间适中,峰形尖锐,可满足对20种目标物的定性、定量检测要求。
比较乙腈-水和甲醇-水两种流动相,结果表明乙腈-水作为流动相时分离度和灵敏度明显优于甲醇-水。为进一步提高化合物的离子化效率,增加灵敏度,结合目前LC-MS/MS法检测雌性激素和孕激素的相关报道[7,15],在流动相中添加对负离子有增强作用的氨水,通过比较0.01%~0.04%的氨水浓度对灵敏度的影响,最终确定流动相为乙腈-0.02%氨水溶液;再优化流动相比例,最终选择线性梯度洗脱,目标物分离度较好、保留时间适中,优化后的多反应监测总离子流色谱图见图1。
2.3 表面修饰MWCNTs的表征
本实验参考彭晓俊等[16]对MWCNTs的表面改性处理方式,用浓H2SO4和浓HNO3组成的混酸对MWCNTs进行化学切割,并在碳纳米管的表面氧化形成一定量的含氧官能基团-COOH和-OH。本实验与彭晓俊等的工艺不同,增加浓HNO3的比例,选择120 ℃回流1 h,再60 ℃回流12 h,可有效除去MWCNTs中的杂质颗粒,增加MWCNTs表面含氧官能团的数量。氧化修饰后MWCNTs可以在水中分散成悬浮液,黑色颗粒没有下沉,这可能是由于氧化修饰后的MWCNTs中含有-COOH等含氧亲水官能团,增加了MWCNTs的亲水性[17]。应用扫描电镜进行表征,结果如图2所示,氧化处理后的MWCNTs长度分布均匀,长径比适中,其傅里叶变换红外(FT-IR)光谱测试的结果与彭晓俊等[16]的结果相同,在特征峰3 734 cm-1出现羧基的特征峰,在3 414 cm-1处出现羟基的伸缩振动峰,因此表明对MWCNTs的表面氧化修饰产生了羟基和羧基官能团。

图2 表面修饰MWCNTs的扫描电镜(SEM)图Fig.2 SEM images of surface modified MWCNTs
2.4 样品提取方式的优化
牛奶成分复杂,除含大量水外还含有蛋白质、脂肪、碳水化合物等成分。牛奶中农兽药残留的检测大多采用乙腈作为提取溶剂。本文分别考察了甲醇、乙腈、叔丁基甲醚和乙酸乙酯4种提取溶剂的提取效果,结果与曹慧关于牛奶中青霉素的检测[18]和赵超敏关于牛奶中4中雌性激素的检测[19]的研究结果一致,甲醇和乙腈都具有提取雌激素的同时沉淀蛋白质的作用,而乙腈沉淀蛋白的能力较甲醇强,沉淀效果好,离心后上清液与沉淀物完全分离,故本研究采用乙腈作为提取溶剂。
2.5 固相萃取条件的优化
表面修饰MWCNTs作为固相萃取柱的填料是影响净化效果的直接因素,为了确保固相萃取小柱填料的均匀性,减少萃取柱中气泡,本实验采用湿法装柱,与彭晓俊等[16]和寇立娟等[20]采用的干法填充相比,表面氧化修饰MWCNTs填充的更加均匀、紧密,净化效果更好。本实验先将混合标准溶液添加至5 mL 乙腈-水(2∶1,V/V)的溶液中,确定提取液的稀释倍数,再将混合标准溶液添加至空白样品中,研究淋洗液和洗脱液的种类、用量等因素对固相萃取的影响,考察固相萃取参数。
2.5.1提取液稀释倍数的确定移取500 μL混合标准储备液(10 mg/L)至50 mL乙腈-水(2∶1,V/V)溶液中,分别移取5 mL加标溶液5份,采用不稀释、5倍稀释、10倍稀释、15倍稀释、45 ℃氮气吹干后用水复溶的5种方式经固相萃取柱净化,通过检测流出液中目标化合物的浓度,考察乙腈提取液的处理方式,结果表明:提取液不经稀释直接过固相萃取时,流出液中可检测出7种雌性激素,浓度在40~60 μg/L之间,这是由于雌性激素在乙腈中的溶解性好,分配系数大导致目标化合物没有被固相萃取柱全部吸附;采用将提取液45 ℃氮气吹干再用水复溶后过柱的净化方式,由于提取液中含有大量水,浓缩速度慢、耗时长,且浓缩过程中会产生悬浮固形物,堵塞柱筛板,造成部分目标化合物的损失。将提取液用水稀释10倍后过柱,流出液中7种目标化合物均未检出,表明溶液中乙腈含量低于7%时,7种雌性激素可以被氧化修饰MWCNTs固相萃取柱全部吸附。因此本实验选择将提取液用水稀释10倍后再使用固相萃取柱净化。

图3 样品溶液的pH对萃取效率的影响Fig 3 Effect of the pH value of the sample solution on the extraction efficiency
2.5.2溶液pH值的影响样品溶液的pH值能影响化合物的电离平衡,改变目标分析物存在状态,影响目标化合物与萃取剂之间的分配系数[21],还能改变固相萃取材料氧化修饰MWCNTs表面官能团的类型和密度,影响对目标化合物的吸附效果[22]。本文所研究的目标化合物呈弱酸性,所以考察pH值为2~10时MWCNTs固相萃取柱对目标化合物的萃取效果,选择pH值影响较大的4种雌性激素的回收率结果绘制图3,结果表明萃取效率随着pH值的升高而降低,特别是在碱性条件下,萃取效率急速下降,溶液的pH=6.0±1.0时的萃取效果较好。故纯牛奶样品可以直接采用纯水稀释后上样,而酸奶或乳酸饮料等特殊样品需采用纯水稀释后再调节pH=6.0±1.0后上样。
2.5.3淋洗液和洗脱液的选择及用量选择合适的淋洗液可有效去除未被吸附的杂质和被吸附的极性较大杂质,减小给质谱分析带来的基质效应。分别采用5%、10%、15%、20%的甲醇溶液作为淋洗液进行试验,研究结果表明采用10 mL 10%的甲醇溶液作为淋洗液时各化合物的基质效应低,且淋洗液中7种雌性激素均未检出,故采用10 mL水和10 mL 10%的甲醇溶液作为淋洗液,以去除杂质。
本文选择在甲醇和乙腈中添加不同比例的氨水作为洗脱剂,考察洗脱效果。结果表明,5%氨水-甲醇和5%氨水-乙腈的洗脱效果相当,各化合物的绝对回收率均大于80%,而乙腈较甲醇的毒性强,且不宜吹干,所以选择氨水-甲醇(5∶95,V/V)溶液作为洗脱液,分两次进行洗脱,可以达到好的洗脱效果。
2.6 方法学验证
2.6.1线性范围、检出限和定量限分别准确移取混合标准工作液和混合同位内标工作液,用初始流动相配制成1~100 μg/L的系列标准溶液,以雌酮-D2作为雌二醇、雌三醇、雌酮、炔雌醇的同位素内标,己二烯雌酚-D2作为己二烯雌酚的同位素内标,己烷雌酚-D4作为己烷雌酚的同位素内标,己烯雌酚-D8作为己烯雌酚的同位素内标,以待测物质的峰面积和相应内标的峰面积的比值(Y)为纵坐标,质量浓度(X)为横坐标绘制标准曲线,线性回归方程及相关系数见表2。结果表明7种雌性激素的线性关系良好,相关系数均大于0.99;在5 g空白基质样品中加入200 μL混合标准工作液(10 μg/L),理论加标浓度为0.4 μg/kg,按照本文建立分析方法进行检测,结果信噪比均大于100,鉴于质谱本身的稳定性、不同仪器间的差异、信噪比和方法的适用性,确定本方法的检出限为0.02~0.1 μg/kg,定量限为0.06~0.3 μg/kg。

表2 雌性激素的标准曲线、线性范围、相关系数、检出限(LODs)和定量限(LOQs)Table 2 The linear equations,correlation coefficients(r),LODs and LOQs of estrogens
2.6.2方法的适用性、回收率和精密度为了验证方法的准确性和可靠性,在阴性样品中分别加入混合标准溶液和混合同位素内标工作液,使加标水平为0.5、5和10 μg/kg,每个浓度水平按照优化后的方法平行测试6次,回收率和精密度的计算结果见表3。雌性激素的回收率在86.7%~103.6%之间,相对标准偏差(RSD)在1.5%~6.9%之间,表明本方法的准确度和精密度良好。

表3 方法的精密度和回收率(n=6)Table 3 Precision and recovery of the method(n=6)
实际样品和加标样品的色谱图见图4。
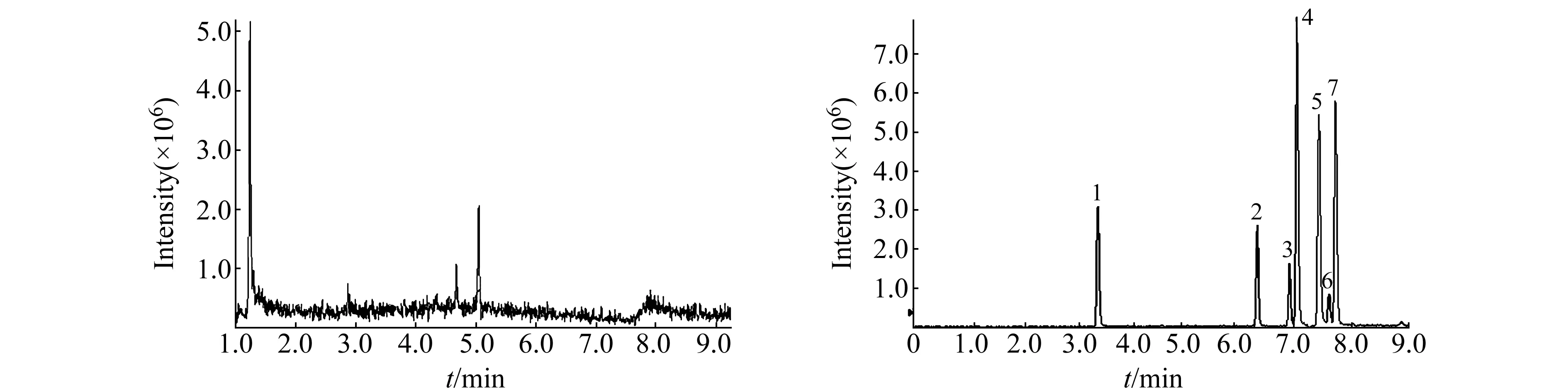
图4 阴性样品(a)及阴性样品中添加7种雌性激素(b)的MRM色谱图Fig.4 MRM chromatograms of a negative milk sample(a) and a negative milk sample spiked with 7 estrogens 1.estradiol;2.estriol;3.ethinylestradiol;4.estrone;5.diethylstilbestrol;6.dienestrol;7.hexestrol.
3 结论
本文采用物理和化学手段对MWCNTs表面进行氧化修饰,采用湿法装柱的方式自制固相萃取小柱,考察溶液的pH值,淋洗液和洗脱液的种类和用量,确定固相萃取参数,实现对牛奶中7种雌性激素的快速富集和净化,结合超高效液相色谱-串联质谱(UPLC-MS/MS)技术,建立了一种同时快速检测牛奶中7种雌性激素残留的分析方法。在样品前处理中加入同位素内标,不但能有效的去除基质效应的影响,还能排除前处理过程因样品损失造成的回收率不稳定的问题。将本文建立的方法与国家标准方法和已报道的方法进行比对,表明该方法检测成本低、分析速度快、基质干扰小、检出限低、定量结果更加准确,为牛奶中雌性激素的微量分析提供更加准确可靠的定性筛查和定量分析方法。
参考文献:
[1] Andringam F A,Frank J C M,Fernandez E,et al.Journal of Veterinary Science,2013,14(2):161.
[2] QIN L Q,WANG P Y,KANEKO T,et al.Medical Hypotheses,2004,62(1):133.
[3] LIN W X,DONG W F,CHEN X,et al.Chinese Journal of Chromatography(林维宣,董伟峰,陈溪,等.色谱),2009,27(3):294.
[4] LI Y,LIU JL,ZHANG S,et al.Chinese Journal of Analytical Chemistry(李鱼,刘建林,张琛,等.分析化学),2012,40(1):107.
[5] DUAN Y S,WANG B L,AI L F,et al.Chinese Journal of Chromatography(段永生,王炳玲,艾连峰,等.色谱),2014,32(6):647.
[6] MENG X,XIE S H,XIE S T,et al.Journal of Analytical Science(孟霞,谢世红,谢世涛,等.分析科学学报),2015,31(2):208.
[7] ZHU W X,LIU Y F,YUAN P,et al.Chinese Journal of Chromatography(祝伟霞,刘亚风,袁萍,等.色谱),2010,28(11):1031.
[8] Iijima S.Nature,1991,354(7):56.
[9] Ren X M,Chen C L,Nagatsu M,et al.Chemical Engineering Journal,2011,170(s2-3):395.
[10] Hu C,Chen B,He M,et al.Journal of Chromatography A,2013,1300:165.
[11] Asensio-Ramos R M,Dorazio G,Hernandez-Borges J,et al.Analytical and Bioanalytical Chemistry,2011,400(4):1113.
[12] ZHAO H X,SHI W L,SUN D J,et al.Chinese Journal of Analytical Chemistry(赵海香,史文礼,孙大江,等.分析化学),2009,37(10):1479.
[13] Tan C J,Chua H G,Ker K H,et al.Analytical Chemistry,2008,80(3):683.
[14] Valcarrcel M,Cardenas S,Simonet B M.Analytical Chemistry,2007,79(13):4788.
[15] YAO W X,YING J B,ZHANG S L.et al.Chinese Journal of Chromatography(姚伟宣,应剑波,张素玲,等.色谱),2015,33(4):342.
[16] PENG X J,PANG J S,DENG A H,et al.Chinese Journal of Chromatography(彭晓俊,庞晋山,邓爱华,等.色谱),2012,30(9):966.
[17] ZHU S K,SU X L.Journal of Analytical Science(朱书奎,粟学俐.分析科学学报),2012,28(6):775.
[18] CAO H,CHEN X Z,ZHU Y,et al.Journal of Chinese Mass Spectrometry Society(曹慧,陈小珍,朱岩,等.质谱学报),2015,36(1):23.
[19] ZHAO C M,YUE Z F,WU J,et al.Modern Food Science and Technology(赵超敏,岳振峰,吴军,等.现代食品科技),2014,30(6):244.
[20] KOU L J,LIANG R N.Chinese Journal of Chromatography(寇立娟,梁荣宁.色谱),2014,32(8):817.
[21] LI H F,YANG H Y,ZHANG Y,et al.Chinese Journal of Chromatography(李海芳,杨红云,张英,等.色谱),2014,32(4):413.
[22] Pemg X J,Li Y H,Luan Z K,et al.Chemical Physics Letters,2003,376(1-2):154.
猜你喜欢
口腔护理用品工业(2021年4期)2021-11-02 08:22:54
中国特种设备安全(2021年12期)2021-04-26 14:37:00
中国油脂(2020年3期)2020-04-10 02:08:54
中成药(2018年6期)2018-07-11 03:01:32
无机化学学报(2016年8期)2016-12-06 09:05:14
化学分析计量(2016年1期)2016-03-14 00:35:19
中国粮油学报(2016年5期)2016-01-23 02:45:06
分析测试学报(2015年3期)2016-01-13 06:18:20
水生生物学报(2015年1期)2015-11-05 06:32:38
食品科学(2014年21期)2014-03-08 06:13:56
