
液相色谱-串联质谱法同时测定油茶不同组织中5类内源激素
2018-05-07 02:42阮成江李景滨
分析科学学报 2018年1期
杜 维, 阮成江*, 丁 健, 李景滨, 李 贺, 王 莉
(生物技术与资源利用教育部重点实验室,大连民族大学资源植物研究所,辽宁大连 116600)
油茶(Camelliaoleifera)是我国南方重要的油料树种,也是世界四大木本油料之一[1]。油茶花果同期,内源激素如生长素(IAA)、赤霉素(GA)、玉米素(tZ)、脱落酸(ABA)和茉莉酸(JA)等会影响果实发育及花芽形成与分化[2 - 4]。前人研究发现,同一结果枝上不同部位激素既相互制约又相互影响,例如油茶果实发育过程中内源GA、IAA和ABA浓度较高[5],但枝条内高浓度GA可能不利于花芽分化与成花[6]。因此同一枝条上不同部位5种内源激素的同时测定有助于探索激素间协同及拮抗作用机理[7 - 8],为阐明内源激素对油茶果实及花芽分化影响以及使用激素诱导油茶花芽分化等研究提供理论依据。
内源植物激素在植物中含量很低,杂质成分多且复杂,难以做到对小范围组织激素的精确测定。目前,对油茶内源激素的测定主要采用酶联免疫法[9]和液相色谱法[10 - 11],还未见使用液-质联用法的相关报道。液-质联用法具有检测器选择性强,灵敏度高等优势[12],但该方法在进样前需要采用固相萃取(SPE)技术去除杂质,以避免质谱污染和减弱杂质带来的基质效应[13 - 17]。由于不同激素间的理化性质不同,不同种类的固相萃取柱往往只能针对2~3种结构相似的激素进行纯化,同时纯化多种激素无法保证较高回收率。本研究充分挖掘混合型阳离子交换SPE柱(MCX)的分离特点,优化提取条件达到只使用单一MCX柱同时对油茶不同组织(枝条、嫩叶、花芽和果实)的IAA、GA4、tZ、ABA和JA 5类激素进行分离纯化,然后利用高效液相色谱-串联质谱法(HPLC-MS/MS)进行测定,为植物中5种内源激素的精确定量提供科学可靠的方法。
1 实验部分
1.1 仪器、试剂与材料
API 3200三重四极杆质谱仪(美国,AB公司);DGU-20A液相色谱系统(日本,岛津公司);MCX固相萃取柱(3 cc/60mg,30 μm,美国Waters公司)。
吲哚-3-乙酸(Indole-3-acetic Acid,IAA)标准品(阿拉丁)、脱落酸(Abscisic Acid,ABA)、茉莉酸(Jasmonic Acid,JA)、玉米素(Trans-zeatin,tZ)、赤霉素(Gibberellin A4,GA4)标准品(美国Sigma公司);甲醇、甲酸(色谱纯,Honeywell);乙酸铵(优级纯,科密欧)。标准溶液配制:分别精确称取2 mg各标准品,溶于5 mL甲醇中,配制成400 μg/mL单标储备液,取5 μL储备液稀释到2 mL为1 μg/mL,用于优化质谱条件。各取250 μL单标储备液用甲醇定容到100 mL,配制成1 μg/mL的标准混合液用于标准曲线的测定,标准混合液浓度梯度为0.5、1.5、10、50、100、500、1 000 ng/mL。
油茶不同组织(二年生枝条、嫩叶、花芽、种仁)样本于2016年9月7日采自贵州玉屏侗族自治县。取样后经液氮冷冻,于-80 ℃保存。
1.2 实验条件
1.2.1液相色谱-质谱条件色谱条件:采用岛津C18色谱柱(150×4.6 mm,5 μm);ESI(+)模式下流动相A为0.1%甲酸,流动相B为甲醇;ESI(-)模式下流动相A为水溶液,流动相B为甲醇。质谱条件:为使目标物尽可能离子化,提高仪器测定灵敏度,采用电喷雾离子源正离子模式(ESI+)检测IAA、tZ;负离子模式(ESI-)检测ABA、GA4、JA。离子化电压(+5 500/-4 500 V);TEM 550 ℃;气帘气压力30 psi;扫描方式为多反应监测(MRM)模式。优化后的质谱条件和液相条件如表1、2所示,质谱图如图1所示(浓度30 ng/mL)。

表1 MRM模式下质谱参数Table 1 MRM parameters for tandem mass spectrometry
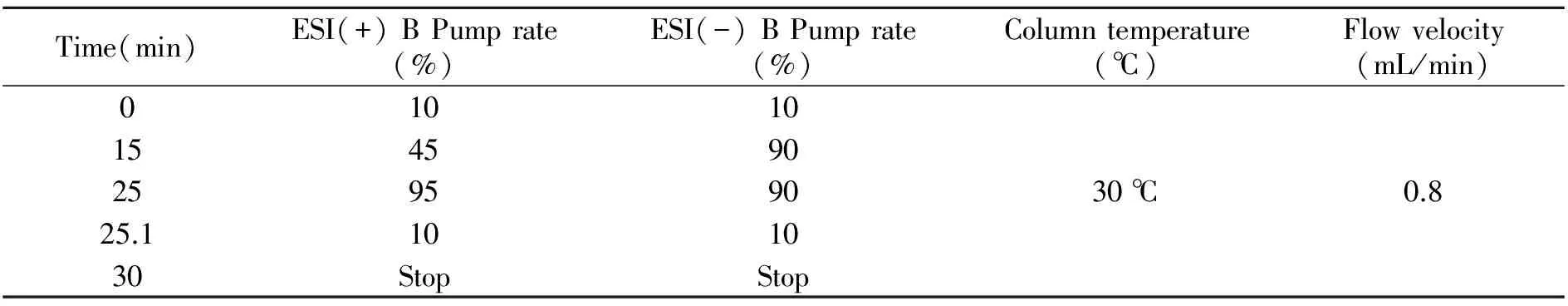
表2 激素测定的高效液相色谱条件Table 2 Conditions of HPLC for measurement of hormones
*:mobile phase A was water,B was methanol.
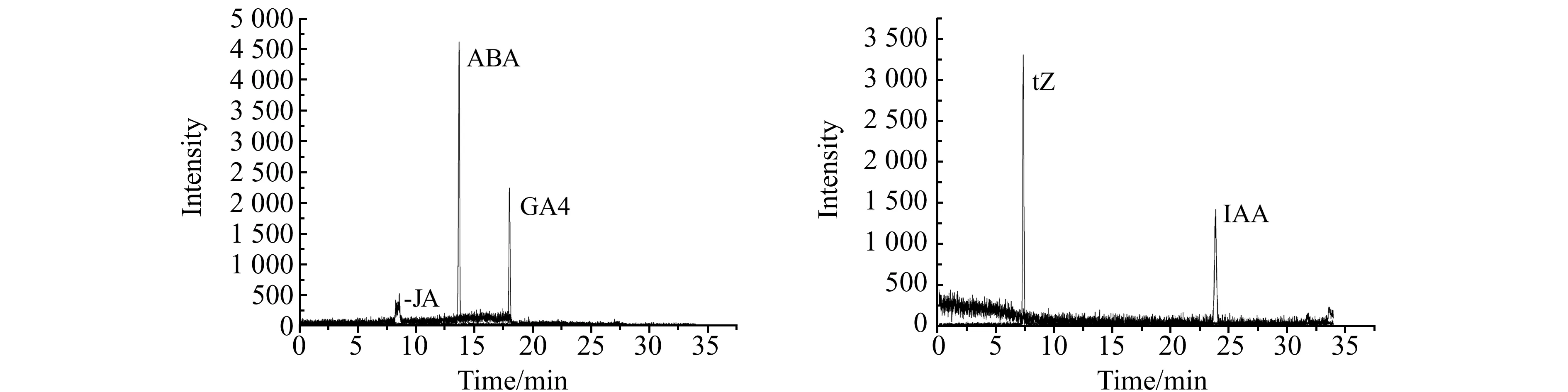
图1 5种被测激素的MRM色谱图Fig.1 MRM chromatograms of five hormone
1.2.2样品处理与条件优化取油茶不同组织样本1~2 g,置于研钵中,加入液氮快速研磨,将磨碎的粉末称重后(差量称重)移入离心管中,加入5 mL提取溶剂(含75%甲醇、5%甲酸水溶液),于4 ℃过夜提取。次日10 000 r/min 离心15 min,上清液转移到新管,沉淀用2 mL提取液清洗两次,合并提取液后用氮气吹干甲醇。
固相萃取前需要将氮吹浓缩后的样品复溶,溶剂极性较弱会影响非极性样品与固相萃取柱的结合效率,导致样品回收率降低;溶剂极性过强可能会使非极性样品复溶效率降低。为了保证固相萃取最佳的提取效率和回收率,对固相萃取前激素复溶的溶剂极性进行优化。用含有不同浓度甲醇的1 mol/L甲酸水溶液复溶,定容至10 mL,进行固相萃取。
1.2.3固相萃取与条件优化固相萃取柱按填料种类划分可分为正相材料、反相材料、无保留载体材料。除无保留载体材料外,正反相材料的保留机理大多包含极性/非极性相互作用、离子交换作用。本文采用混合阳离子反向交换柱(MCX)作为固相萃取柱,MCX同时具有强阳离子交换作用、非极性相互作用以及弱极性相互作用。分别用2 mL甲醇、2 mL 1%甲酸水溶液活化,之后加入样品溶液过柱,甲醇洗脱得到IAA、GA4、ABA、JA;用含有0.35 mol/L氨水的60%甲醇水溶液洗脱得到tZ,在保证回收率前提下以1、1.5、2、3 mL梯度对激素洗脱体积进行优化。混合两次洗脱液氮气吹干后用甲醇定容,过0.22 μm滤膜后,待上样测定。
1.2.4方法学考察稀释标准品混合液,配制成6个浓度梯度混合标准品溶液(1、5、20、50、100、200、500 ng/mL),测定后绘制标准曲线。取不同数量级的2个浓度(1 ng/mL和100 ng/mL)标准混合液,待样品粉碎后加入,考察方法的精密度以及标准品在叶片样品中加标回收率,每组实验做三组平行。以三倍信噪比(S/N=3)计算检出限。
2 结果与讨论
2.1 激素提取原理
由于植物样品中含有大量天然吡咯衍生物类、多烯类、酚类和酮类色素,如不去除会对色谱柱和离子源造成污染。本文用非极性溶剂提取,极性溶剂复溶的方法可以除掉大部分的吡咯衍生物类、多烯类以及酮类色素,后经固相萃取可以去除一部分的酚类色素。内源激素IAA、ABA、GA4、JA和tZ的logP值(亲水系数)分别为2.26、2.24、2.13、2.02和0.776,疏水性较强的前4种激素(酸性激素)在强酸性条件下不电离,可用非极性相互作用在MCX萃取柱中保留,用甲醇即可洗脱。极性较强的碱性激素tZ在酸性条件下带正电荷被MCX以更强的阳离子交换作用所保留,可用0.35 mol/L 60%的氨化甲醇洗脱,这样可做到只使用MCX一种固相萃取柱即可对这5种激素进行保留和洗脱。
2.2 固相萃取前溶剂极性优化
对固相萃取前溶剂极性优化(图2),以强阳离子交换作用得到保留的tZ回收率一直保持在95%以上,除tZ外溶剂极性对其他激素回收率的影响较大。随着溶液极性的下降可以增加非极性激素的溶解性,但会显著降低固相萃取柱对非极性相互作用四种激素(IAA、GA4、ABA和JA)的保留。在不含甲醇的溶剂下该四种激素提取率达到最高的97%、99%、95%和89%。故选用1 mol/L甲酸溶液(不含甲醇)复溶提取物可以保证目标激素有较高的回收。
2.3 固相萃取洗脱体积的优化
保证回收率前提下对激素洗脱体积进行优化(图3),由于tZ为酸性上样/碱性洗脱,洗脱体积对回收率影响较大,3 mL的洗脱体积才能保证将95%以上的样品洗脱下来,其余激素2 mL的洗脱液能保证有较高的回收率。洗脱条件确定为:2 mL甲醇洗脱得到IAA、GA4、ABA、JA,3 mL含0.35 mol/L氨水的60%甲醇水溶液洗脱得到tZ。
2.4 标准曲线、检出限与定量限
在实际样品中加入标准品绘制标准曲线,可以去除基质效应对实验准确性的影响。由于很难找到不含激素的植物样品,在实际样品中加入标准品会使标准曲线的低浓度范围无法考察,使整个实验的线性范围偏高,故本实验采用标准品考察线性方程,采用加标回收率考察基质效应。IAA、ABA、GA4、tZ和JA 5种激素标准曲线的线性相关系数R2均大于0.995(表3),呈高度线性;按3倍信噪比(S/N=3)计算检出限,换算浓度后得出检出限依次为0.7、1.2、0.8、0.4、0.5 ng/g,根据植物的一般激素水平(0.1~100 ng/g),每次样品量只需要0.5 g,有利于对植物细小位置内源激素的精确测定。
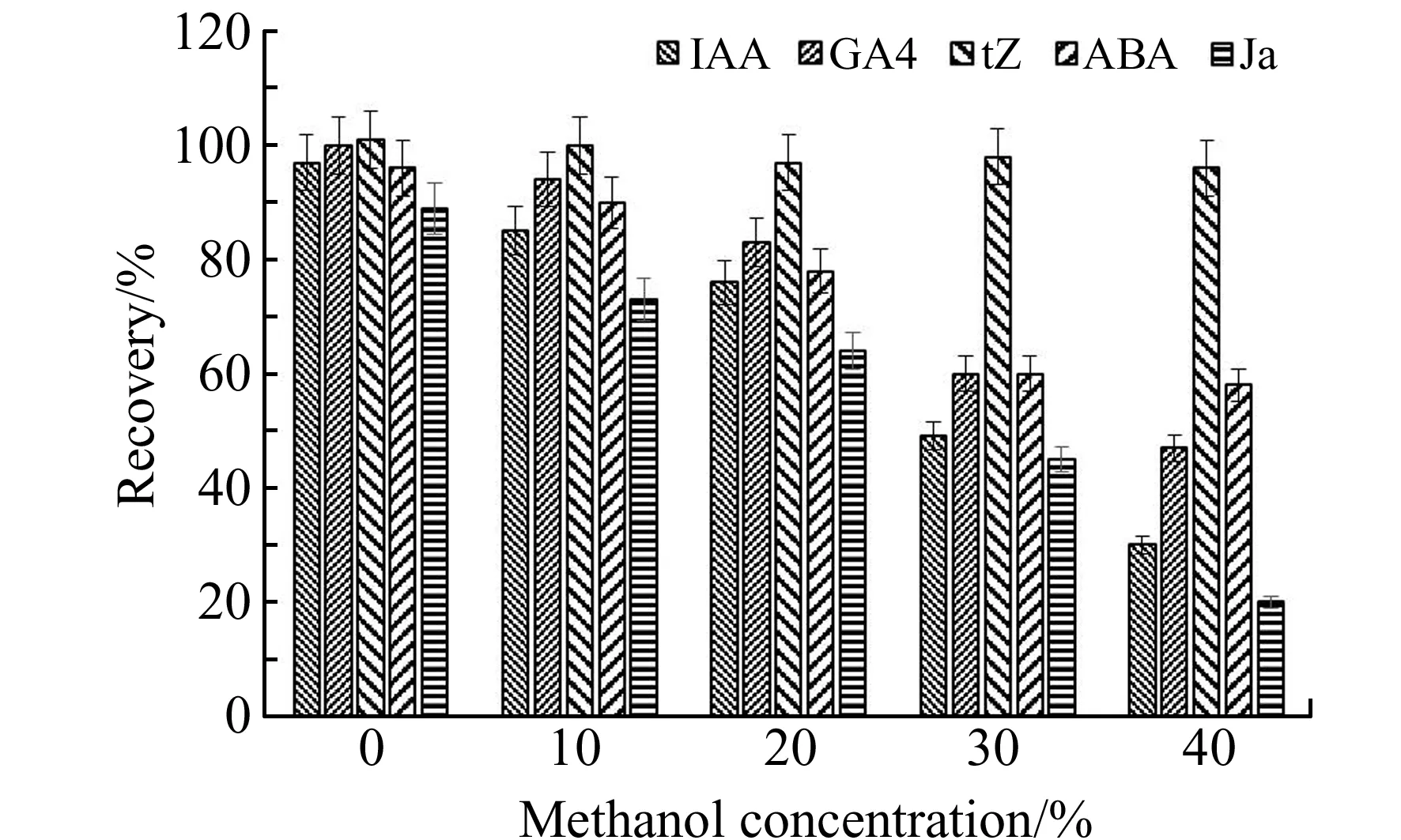
图2 溶剂极性对激素回收率影响Fig.2 The solvent polarity effect on hormone recovery

图3 洗脱体积对激素回收率影响Fig.3 The solvent volume effect on hormone recovery

表3 线性方程、检出限与定量限Table 3 Linear equation,limit of detaction (LOD) and quantification of detection (QOD)
2.5 方法精密度、回收率与基质效应
实验结果表明,5种激素测定的日内精密度(RSD)在5.2%以下(表4),日间RSD在8%以下,回收率大于80%,标准偏差(SD)在10%以内,方法稳定,误差较小。
基质效应在液-质联用主要体现在样品中的杂质会影响被测物的离子化程度,内源性的组分如磷脂等会对ESI离子源中被测物电离产生比较强的抑制作用。评价基质效应的强弱一般用不含被测物成分的样品加入被测物标准品进行测定,与预期结果相比观察测定数据的变化。由于很难找到不含激素的植物样品,本实验向提取液中加入已知浓度标准品来确定基质效应的强弱。实验结果表明,5种内源激素加标回收率在79%~101%间,<25%的基质效应显著值,基质效应不明显,在实测样品时可根据加标回收率对实测值进行相应补偿。
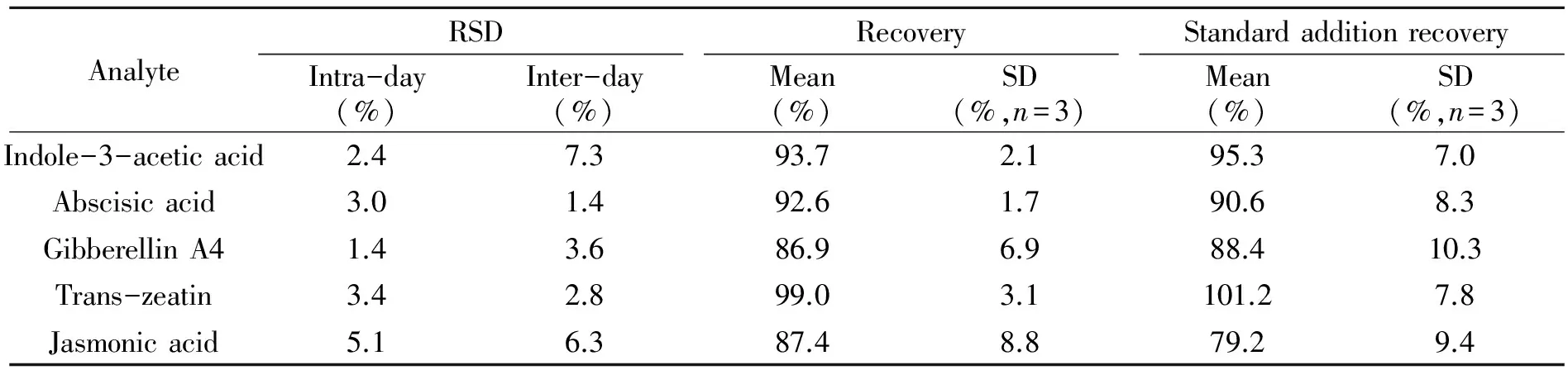
表4 精密度、回收率与基质效应Table 4 Relative standard deviation (RSD),recovery and standard addition recovery
2.6 方法应用
用本实验建立的方法同时测定油茶不同组织中的5种内源激素,结果表明:(1)生长素在花芽中含量最高为1821.6 ng/g,其次为嫩叶218.5 ng/g,但在种仁中较低,仅为57.3 ng/g,且在枝条中未被检出。9月7日为油茶果实生长发育的末期,其种仁生长素含量低,花芽处于旺盛期(10月为花期),生长素含量较高;在花果同期的时间段,油茶枝条生长停止,主要养分用于花芽生长、维持嫩叶生长和种仁油脂积累,这与生长素在不同组织的分布规律相同。(2)赤霉素A4在嫩叶、种仁官内含量较高分别为227.6、52 ng/g,但在花芽中赤霉素A4含量较低(16.5 ng/g),在枝条中未被检出。赤霉素在嫩叶种仁等细胞分裂旺盛组织内含量较高,与其具有促进植物生长和开花等作用的结论相一致。(3)玉米素是细胞分裂素的一种,在油茶组织内与其他激素相比含量最低,在嫩叶、种仁和枝条中含量较花芽高,分别为3.6、2.3、2.3 ng/g,花芽中玉米素含量最低为1.5 ng/g,分布规律不明显。(4)脱落酸在花芽和枝条中含量较高,分别为445.2、456.6 ng/g,是生长旺盛的组织(嫩叶和种仁)含量的3~4倍。油茶花芽从形成至开花需要5~6个月,生长较慢可能与花芽内高含量ABA有关。(5)茉莉酸在嫩叶、花芽和枝条中含量接近,在种仁内达到最高的978.3 ng/g,是其他组织的2倍以上。种仁作为油茶油脂积累组织,组织内含有丰富亚油酸和亚麻酸是JA的合成原料,合成的JA同时又会促进种仁的发育,这可能是种仁高JA含量的原因。(6)GA与ABA是互为拮抗作用的两种激素,GA/ABA比值的高低体现出植物组织的生长速度的快慢。测定结果中GA4/ABA比值由高到低依次为嫩叶、种仁、花芽以及枝条,与油茶该部位的实际分生速度相符。

表5 油茶不同组织中激素测定结果Table 5 Determination results of hormones in different organs of oil-tea
ND:not detected.
3 结论
本文建立了只用单一MCX固相萃取柱,结合液-质联用技术同时分析油茶枝条、嫩叶、花芽和种仁中IAA、GA4、tZ、ABA和JA 5种内源激素的定量方法。优化了固相萃取条件,解决了多种激素同时测定回收率较低的问题。测定结果显示油茶的花芽和嫩叶的生长素水平较枝条和果实高,种子中茉莉酸含量最高(978.3 ng/g),GA4/ABA比例为嫩叶>种仁>花芽>枝条,与油茶各部位生长速度一致。该方法简便快速,与已发表酶联免疫法和液相色谱法内源激素测定文献相比需要的样品量少(100~500 mg)、精密度高(RSD<7%),可以作为一种高通量快速测定植物内源激素的方法。
参考文献:
[1] CHENG H,LI L L,YUAN H H,et al.Northern Horticulture(程华,李琳玲,袁红慧,等.北方园艺),2013,22(11):5.
[2] MO C M,TU D P,HUANG J,et al.Acta Botanica Boreali-Occidentalia Sinica(莫长明,涂冬萍,黄杰,等.西北植物学报),2015,35(1):98.
[3] Duclos D V,Thomas B.Journal of the American Society for Horticultural Science,2015,140(1),57.
[4] GAO A L,LI J A,LIU R,et al.Nonwood Forest Research(郜爱玲,李建安,刘儒,等.经济林研究),2008,28(2):131.
[5] HU Y L,YAO X H,ZHANG S,et al.Acta Agriculturae Universitatis Jiangxiensis(胡玉玲,姚小华,张山,等.江西农业大学学报),2016,38(3):412.
[6] CHEN X.The Study of Flowering Mechanism of Camellia Oleifera Regulated by Exogenous Gibberellin.Chang Sha:Central South University of Forestry and Technology(陈显.外源赤霉素对油茶成花调控机理的研究.长沙:中南林业科技大学),2013.
[7] YIN P P,BAO R S,DAI J K,et al.Acta Agriculturae Jiangxi(尹培培,包日双,戴佳锟,等.江西农业学报),2012,24(6):37.
[8] DUAN Na,JIA Y K,Xu J,et al.Chinese Agricultural Science Bulletin(段娜,贾玉奎,徐军,等.中国农学通报),2015,31(2):159.
[9] CAO Y Q,YAO X H,REN H D,et al.Journal of Beijing Forestry University(曹永庆,姚小华,任华东,等.北京林业大学学报),2015,37(11):76.
[10] CHEN B W,LIU H L,CHEN X M,et al.Shandong Agricultural Sciences(陈博雯,刘海龙,陈晓明,等.山东农业科学),2012,44(3):105.
[11] ZHOU Y M,QI X.Food Science(周艳明,忻雪.食品科学),2010,31(18):301.
[12] Liu H B,Li X H,Xiao J H.Plant Methods.2012.8(1):2.
[13] WANG L,WU Q,DUAN C F,et al.Chinese Journal of Chromatography(王璐,吴倩,段春凤,等.色谱),2011,29(9):923.
[14] WANG Y L,WANG P,WANG C Y.Journal of Analytical Science(王水良,王平,王趁义.分析科学学报),2010,26(5):547.
[15] ZHONG D L,DING M,TANG F B,et al.Chinese Journal of Analytical Chemistry(钟冬莲,丁明,汤富彬,等.分析化学),2013,41(11):1739.
[16] Alder L,Luderitz S,Lindtner K,et al.Journal of Chromatography A,2004,1058(2):67.
[17] Hajslova J,Zrostlikova J.Journal of Chromatography A,2003,1000(1):181.
猜你喜欢
东坡赤壁诗词(2022年4期)2022-10-30
河北果树(2021年2期)2021-04-24
经济林研究(2021年1期)2021-04-10
中国粮油学报(2018年12期)2018-03-19
益寿宝典(2018年17期)2018-01-26
新疆农业科学(2017年4期)2017-06-08
星星·散文诗(2017年34期)2017-02-17
上海农业学报(2016年2期)2016-10-27
华东理工大学学报(自然科学版)(2015年4期)2015-12-01
动物营养学报(2015年10期)2015-12-01
