
大体积样品堆积-微乳液胶束电动色谱法测定5种黄酮类化合物
2018-05-07 00:40齐烨迪林诗瑶林振宇余丽双
分析科学学报 2018年1期
苏 慧, 齐烨迪, 林诗瑶, 赖 昕, 陈 莉,林振宇, 余丽双*,
(1.福建中医药大学药学院,福建福州 350122;2.福州大学食品安全分析与检测教育部重点实验室,福建福州 350116)
黄酮类化合物是中草药中普遍存在的有效成分,它是具有抗肿瘤、抗病毒、解痉抑菌、消炎止咳等生理活性的化合物[1]。现有的黄酮类化合物的检测方法主要有高效液相色谱法(HPLC)[2 - 5]和毛细管电泳法(CE)[6 - 9]。CE具有高效、快速、经济环保等优点,已报道应用于分离检测黄酮类化合物的毛细管电泳分离模式有毛细管区带电泳(CZE)[6]﹑胶束电动色谱(MEKC)[7]、毛细管电色谱(CEC)[8]和微乳液胶束电动色谱(MEEKC)[9]。目前,MEEKC已被广泛应用于分析天然产物[10 - 11]和环境样品[12]等复杂样品。但现有MEEKC法多使用紫外检测器,灵敏度低,限制了其在痕量分析中的应用。
为了提高分析方法的灵敏度,可将MEEKC法同在线富集技术联用,目前常用的在线富集方法有常规样品堆积法、大体积样品堆积法和场放大样品堆积法等。其中,常规样品堆积法富集效果较低,场放大样品堆积法只适合富集低盐和无盐样品,而对于尿液等复杂高盐样品,富集效果不明显。大体积样品堆积法(LVSS)是通过压力进样,使样品溶液充满5%~60%的毛细管,然后施加反向电压将样品基体不断泵出管外以减小其基质干扰,并产生样品堆积作用,以提高检测灵敏度[10,13 - 14]。 因此,大体积样品堆积-微乳液胶束电动色谱(LVSS-MEEKC),适用于复杂生物样品的痕量分析[10,14]。 至今尚未见利用LVSS-MEEKC同时分离检测表儿茶素、槲皮苷、槲皮素、木犀草素、山奈酚5种黄酮类化合物的报道。本文采用LVSS-MEEKC法分离检测表儿茶素、槲皮苷、槲皮素、木犀草素、山奈酚5种黄酮类化合物,结果表明方法富集效果良好,检测限可低至ng/mL。建立的新方法成功用于人尿加标样品中上述5种黄酮类化合物的检测。
1 实验部分
1.1 仪器与试剂
Beckman PA800plus型毛细管电泳仪(美国,Beckman Coulter公司),配二极管阵列检测器及32Karat软件;弹性未涂层石英毛细管(60 cm×75 μm,有效长度49 cm,永丰县锐沣色谱器件有限公司);KQ-500DE型数控超声波清洗器(昆山市超声仪器有限公司);QL-861型涡旋仪(江苏海门市其林贝尔仪器制造有限公司);Starter 3C型pH酸度计(上海精密科学仪器有限公司);XS105型电子分析天平(梅特勒托利普国际股份有限公司)。
表儿茶素、槲皮苷、槲皮素对照品均购自中国食品药品检定研究院;山奈酚对照品购自成都曼思特生物科技有限公司;木犀草素对照品购自Sigma生物制品有限公司。对照品纯度均≥98%。对照品溶液:分别精密称取表儿茶素、槲皮苷、槲皮素、木犀草素、山奈酚适量,加入无水乙醇溶解,配制成浓度为1 mg/mL的对照品储备液。十二烷基硫酸钠(SDS,Amresco);无水乙醇、乙酸乙酯和正丁醇为色谱纯试剂(上海阿拉丁试剂有限公司);硼砂和NaH2PO4等试剂为分析纯试剂,聚酰胺(30~60目)(国药集团化学试剂有限公司)。所有水均经Milli-Q超纯水系统(Millipore公司)处理。
尿液来源于实验室成年男性志愿者,均为未服用任何药物的空白尿液。
1.2 样品处理
往空白人尿样中分别加入高、中、低三种浓度水平的5种黄酮类化合物的混合对照品溶液,浓度分别为:表儿茶素0.08、0.1和0.12 μg/mL,槲皮苷、山奈酚、槲皮素、木犀草素均为0.04、0.05和0.06 μg/mL。随后,将加标尿样置于离心机中,以2 000 r/min离心5 min。收集上清液,供LVSS -MEEKC分析。
1.3 电泳工作条件
新毛细管使用前分别用水、0.1 mol/L HCl、水、0.1 mol/L NaOH溶液、水各冲洗20 min。旧毛细管使用前分别用水、0.1 mol/L NaOH溶液、水、运行缓冲溶液各冲洗8 min。MEEKC运行缓冲溶液为:20 mmol/L硼砂-40 mmol/L NaH2PO4(pH=9.0)+0.3%SDS+2%正丁醇+1%乙酸乙酯。每次进样前,毛细管需用运行缓冲溶液冲洗3 min。运行电压:20 kV,检测波长:214 nm,压力进样:20.7 kPa×55 s,堆积时间:1.25 min,操作温度:25 ℃。
1.4 LVSS富集条件
LVSS最佳富集条件为:分离电压20 kV,进样时间:20.7 kPa×55 s,堆积时间:1.25 min,堆积电压:-20 kV。
2 结果与讨论
2.1 MEEKC分离条件的优化
2.1.1缓冲溶液的pH值优化缓冲溶液的pH值对实验结果起着至关重要的作用。运行缓冲液的pH值影响电渗流(EOF)及其分析物的总带电量。实验中考察了pH在7.0~9.2范围对5种黄酮类化合物分离效果的影响。由图1可知:随着pH值增大,山奈酚与木犀草素的分离度变大,木犀草素与槲皮素的分离度先变小后变大;当pH值大于9.0,则表儿茶素的峰展宽;当pH=9.0时,5种黄酮类化合物分离效果最佳,故选择最佳pH为9.0。
2.1.2表面活性剂SDS浓度优化在5 mmol/L 硼砂-10 mmol/L NaH2PO4(pH=9.0)条件下,对缓冲溶液中表面活性剂SDS含量进行考察。实验结果证明,SDS含量超过0.5%,则系统出峰时间延长,EOF升高,毛细管柱内焦耳热增加明显,严重影响分离效果。实验中考察了SDS含量在0.1%~0.5%范围对分离效果产生的影响。SDS浓度过高,分离时间延长,SDS浓度太低,微乳液体系不稳定。实验最选择是0.3%SDS。
2.1.3其他分离条件优化本实验还对助表面活性剂正丁醇等进行考察。结果表明最佳缓冲溶液为:20 mmol/L硼砂-40 mmol/L NaH2PO4(pH=9.0)+3%SDS+2%正丁醇+1%乙酸乙酯。在以上条件下,5种黄酮类化合物得到很好的分离,见图2(b)。
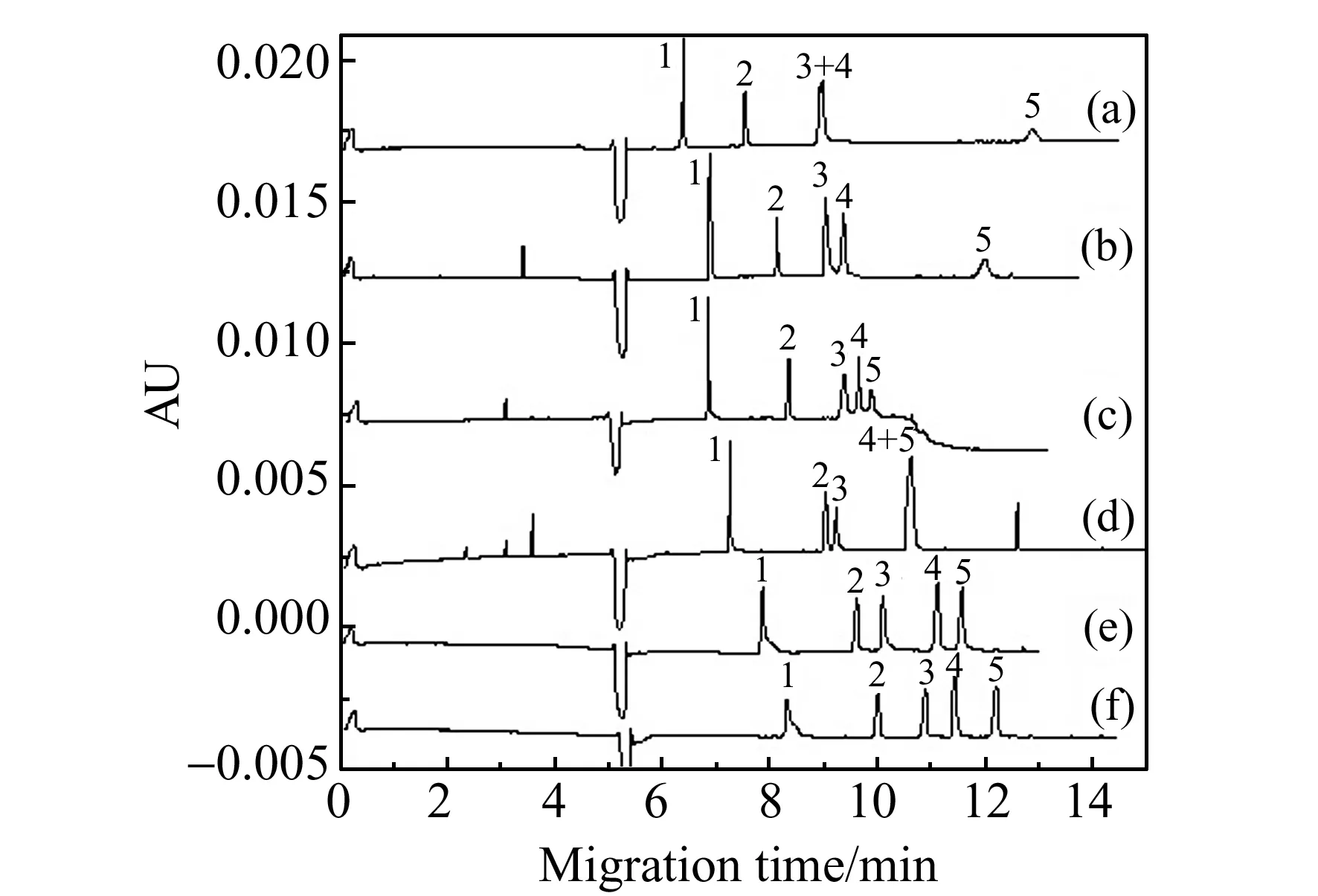
图1 缓冲溶液pH值对分析物迁移行为的影响Fig.1 Effect of buffer pH on migration behavior of analytesRunning buffer:20 mmol/L borate-40 mmol/L phosphate buffer(pH=9.0) with 0.3% SDS,1% ethyl acetate,2% butyl alcohol.separation voltage:20 kV,detection wavelength:214 nm,hydrodynamic injection:3.4 kPa×6 s,temperature:25 ℃.pH:a.7.0;b.7.5;c.8.0;d.8.5;e.9.0;f.9.2.Peak identifications:1.L-Epicatechin;2.Quercitrin;3.Kaempferol;4.Luteolin;5,Quercetin.Sample concentration:10 μg/mL.
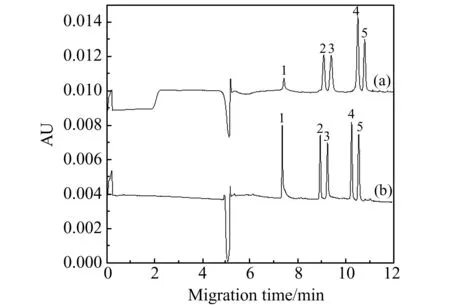
图2 5种分析物的LVSS-MEEKC(a)和MEEKC(b)电泳图Fig.2 Electropherograms of analytes by LVSS-MEEKC(a) and MEEKC(b)Running buffer:20 mmol/L borate-40 mmol/L phosphate buffer(pH=9.0) with 0.3% SDS,1% ethyl acetate,2% butylalcohol.Peak identification:1.L-Epicatechin;2.Quercitrin;3.Kaempferol;4.Luteolin;5,Quercetin.a:The sample concentration is 0.25 μg/mL;hydrodynamic injection:20.7 kPa×55 s;sample stacking time:20 kV×1.25 min;b:The sample concentration of 10 μg/mL;hydrodynamic injection:3.4 kPa×6 s.
2.2 LVSS条件优化
2.2.1堆积时间的优化在无限稀释的样品溶液中,堆积时间控制的是样品基质的排出。堆积时间过长,样品离子会因泵出而损失;堆积时间过短,则富集效果不明显或分离效率降低。当施加堆积电压时,电流值增大到分离电流最大值的90%~99%,应立即停止施加电压以确保最佳富集效果。采用-20 kV的堆积电压,对不同堆积时间(0.9~1.5 min)进行考察。结果表明,当堆积时间小于1.1 min时,分离效率较低。当堆积时间达到1.4 min时,峰1(表儿茶素)消失,说明堆积时间过长,表儿茶素完全损失。进而在1.1~1.4 min之间进行进一步优化,选择1.25 min作为最佳堆积时间。
2.2.2进样条件的优化实验考察了进样时间30~65 s时对样品富集产生的影响。结果表明,当进样时间小于50 s时,峰1的峰高小,富集效果差。随着进样时间的延长,峰高增大,当进样时间大于60 s时,峰高近似不变,而峰形逐渐展宽,导致分离度下降。综合考虑分离度和富集效果,选择进样时间为55 s。
综合上述,最佳LVSS条件为:分离电压20 kV,进样时间:20.7 kPa×55 s,堆积时间:1.25 min,堆积电压:-20 kV。在上述最佳的富集条件下,5种黄酮类化合物富集效果良好(图2)。如图2所示,利用常规MEEKC法测定5种黄酮类化合物(图2(b),分析物浓度为10 μg/mL),其峰面积(除表儿茶素外)与分析物稀释40倍后采用LVSS-MEEKC法(图2(a),分析物浓度为0.25 μg/mL)检测的峰面积相近。以分析物富集前后峰面积比计算富集因子,5种分析物的富集因子在8~59之间,富集效果良好。相比其他4种分析物,表儿茶素富集因子为8较小,可能因为在反向堆积过程中表儿茶素迁移速度较其他4种分析物快,靠近进样端的部分表儿茶素被排出管外,造成损失,导致富集效果下降。
2.3 方法学验证
2.3.1线性关系、定量限和检测限在图2(a)所示实验条件下,对一系列不同浓度的对照品溶液进行LVSS-MEEKC法测定,以各分析物的峰面积(Y)与对应的浓度(X)进行线性回归,所得线性方程、线性范围、相关系数、定量限(S/N=10)和检测限(S/N=3)如表1所示。实验结果表明5种分析物在线性范围内呈良好的线性关系,检测限低至ng/mL级。
2.3.2精密度实验取一定浓度的标准品混合溶液,按图2(a)中所示实验条件连续进样测定5次,计算峰面积和迁移时间的相对标准偏差(RSD),各分析物峰面积和迁移时间RSD均在5%范围内,表明方法的精密度良好。
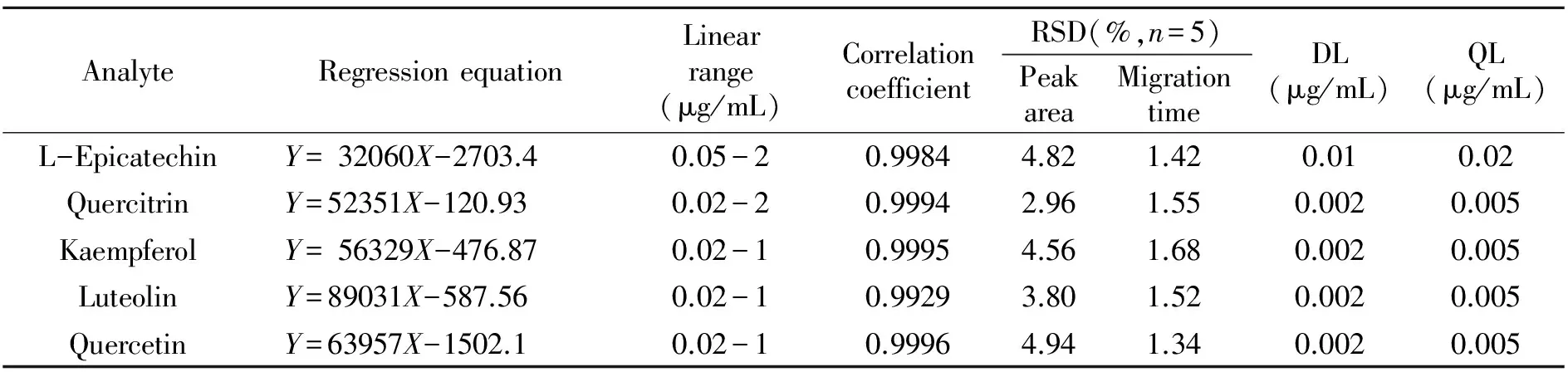
表1 回归方程、线性范围及检测限和定量限Table 1 Regression equation,linear range and detection limit (DLs) and quantitation limit(QLs)
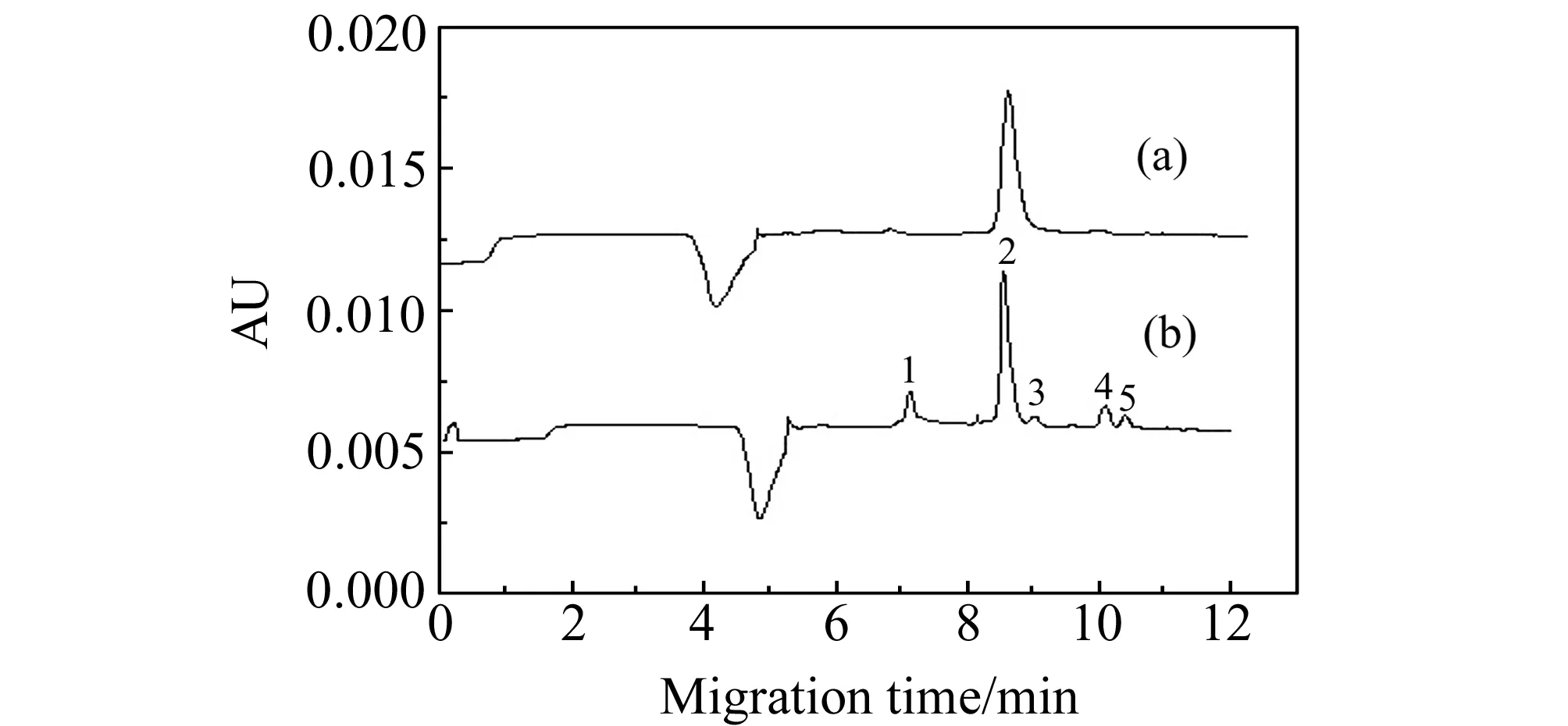
图3 空白尿样(a)及加标尿样(b)的电泳图Fig.3 The electropherogram of human blank urine(a) and spiked urine(b) by LVSS-MEEKC Peak numbers are same to that in Fig.1.
2.3.3加样回收率实验按样品处理方法制备人尿加标样品,在上述最佳富集和分离条件下,进行模拟加标尿样实验,所得谱图见图3,所测得的回收率如表2所示。由图3可知,空白尿样对表儿茶素、槲皮素、木犀草素和山奈酚的测定无干扰,而槲皮苷因无法与空白尿样中的内源性物质峰分离,无法测定。由表2可知,儿茶素、槲皮素、木犀草素和山奈酚4种黄酮类化合物的回收率范围为92.50%~110.0%,RSD小于6.2%。
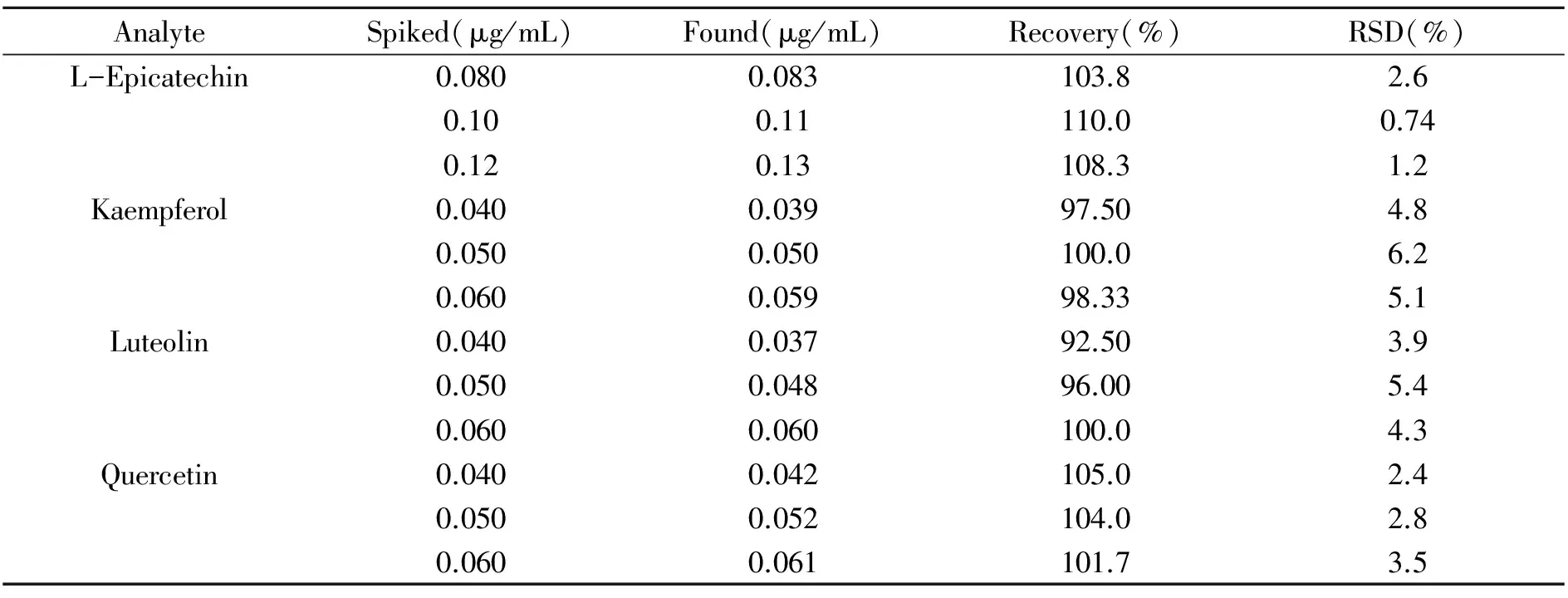
表2 人加标尿样的LVSS-MEEKC加标回收率实验结果(n=3)Table 2 The recovery experiment results of human spiked urine by LVSS-MEEKC(n=3)
3 结论
本文建立的LVSS-MEEKC法可同时分离检测表儿茶素、槲皮苷、槲皮素、木犀草素、山奈酚5种黄酮类化合物。在最优化的富集分离条件下,5种黄酮类化合物在12 min得到基线分离,且分析物的检测限可低至ng/mL级。所建立的新方法可成功用于人加标尿样的测定。
参考文献:
[1] KUANG H X.Chinese Traditional and Herbal Chemistry.Beijing:China Traditional Chinese Medicine Press(匡海学.中药化学.北京:中国中医药出版社),2003:143.
[2] ZHANG L,XIANG C,WANG F,et al.China Journal of Chinese Materia Medica(张雷,向诚,王邠,等.中国中药杂志),2009,34(3):340.
[3] WEI Y J,JIANG J S,TAN X B,et al.Chinese Journal of Modern Applied Pharmacy(韦英杰,姜金生,谭晓斌,等.中国现代应用药学),2012,29(1):60.
[4] YU H,ZHANG X L,XIONG Y Y,et al.Chinese Traditional Patent Medicine(俞浩,张孝林,熊友谊,等.中成药),2014,36(4):830.
[5] SUN Q.Multicomponent Analysis of Thistle Based on Liquid Chromatography and the Pharmacokinetics of Flavonoids.Hebei:Hebei Medical University(孙倩.基于液质联用技术的大小蓟多组分分析与黄酮类成分的药物代谢动力学研究.河北:河北医科大学),2013.
[6] WU T,GUAN Y Q,ZHENG S J,et al.Journal of Analytical Science(吴婷,管月清,郑双杰,等.分析科学学报),2006,22(4):406.
[7] SUN Z X,SU H,CONG R L,et al.Journal of Analytical Science(孙照霞,苏慧,丛日琳,等.分析科学学报) 2016,32(4):541.
[8] LIU H X,LIU F Q,YU A,et al.Chemical Reagents(刘海兴,刘凤芹,于爱民,等.化学试剂),2006,28(6):349.
[9] Yu L S,Xue X Q,Huang L,et al.Electrophoresis,2008,29(3):726.
[10] Yu L S,Lin S Y,Sha M,et al.Analytical Methods,2015,7(22):9489.
[11] Xiao W,Zhang Q,Chen C,et al.Journal Chromatography Science,2016,54(9):1678.
[12] Felici E,Casado C,Wang C C,Electrophoresis,2016,37(22):2977.
[13] Moreno-González D,Lupión-Enríquez I,García-Campaa A M,Electrophoresis,2016,37(22):2963.
[14] CHEN X,NI X J,ZHANG J Y,et al.Chinese Journal of Analytical Chemistry(陈新,倪鑫炯,张佳瑜,等.分析化学),2015,43(1):81.
猜你喜欢
家庭科学·新健康(2022年2期)2022-03-07
天然产物研究与开发(2018年2期)2018-04-04
中成药(2017年12期)2018-01-19
西江月(2017年4期)2017-11-22
小雪花·成长指南(2016年10期)2016-11-01
环球时报(2016-05-18)2016-05-18
中国环境科学(2015年7期)2015-01-28
中国烟草学报(2012年3期)2012-04-10
郑州大学学报(理学版)(2012年4期)2012-03-25
幸福·悦读(2009年8期)2009-12-01
