
不同调节剂制备MOF-Fe的性质及对Seビ的吸附性能
2018-05-05 06:22许海娟魏世勇吴德勇
无机化学学报 2018年5期
王 锐 龚 勇 许海娟 魏世勇*,,3 吴德勇,3
(1湖北民族学院化学与环境工程学院,恩施 445000)
(2湖北职业技术学院,武汉 432000)
(3生物资源保护与利用湖北重点实验室,恩施 445000)
0 引 言
硒是人类必需的微量元素,适量的硒有助于提高人体免疫力等功能,缺硒可引起克山病等疾病,而过量的硒则会导致脱发、指甲变脆、肝增大等中毒症状[1-3]。我国已将硒列为地表水环境质量标准基本检测项目,规定其上限浓度为0.01 mg·L-1[4]。世界上土壤硒的平均含量为0.5 mg·kg-1,我国湖北恩施、陕西紫阳等富硒土壤中硒的含量最高可达1 000 mg·kg-1以上[5]。土壤中的硒可通过矿化、淋溶、生物等作用进入地表水体,玻璃、电池、采矿等行业的含硒废水排放也可导致水体硒含量升高[6]。地表水中硒含量过高是引起动物和人类硒中毒的主要渠道。水体中硒的有效去除方法有共沉淀法、离子交换法、生物方法、吸附等,其中吸附法具有周期短、效率高、无二次污染等优点而被广泛应用[7]。(羟基)氧化铁/铝等金属氧化物比表面积较大、表面活性羟基含量高,被广泛用作水体污染物的吸附剂[7-11]。研究表明,SiO2@氧化铁/氧化铝二元复合物对亚硒酸根(Seビ)的吸附容量分别可达20.4 mg·g-1和 32.7 mg·g-1[10]。 然而,(羟基)氧化铁/铝等金属氧化物对Seビ的吸附容量等性能很难有较大程度的提高[10-11]。因此,有必要展开新材料对Seビ的吸附研究,以获得高效去除水体Seビ的吸附材料,这对水体硒污染的防治具有重要的实践意义。
金属有机骨架材料(MOFs)的比表面积巨大、表面配位不饱和金属位点和官能团丰富,且孔性结构可调,因此被广泛应用于吸附/分离、催化等领域[12-17]。目前,通过向MOFs的合成体系中加入调节剂,研究MOFs形貌/尺寸可控合成及其机理的报道较多。Zhang等[18]研究了酸碱调节剂和配位调节剂对Dy(BTC)(H2O)形貌/尺寸的影响,发现调控MOFs形貌/尺寸的关键因素为合成体系的pH值和配位调节剂。Guo等[19]以不同比例的N,N-二甲基甲酰胺-水(DMF-H2O)混合溶剂作为调节剂,对NH2-MIL-53(Al)形貌/尺寸展开了研究,发现混合溶剂调节剂可以调控NH2-MIL-53(Al)的形貌/尺寸,其调控机理为不同体系中成核速率与生长速率的差异。Liu等[20]研究指出,典型的MIL-101(Fe)形貌为八面体,当调节剂分别为NaAc和甘油时,样品的形貌分别为纺锤体形和双六边形锥体;3种不同形貌的MIL-101(Fe)对 3,3′,5,5′-四甲基联苯胺-H2O2体系的氧化还原性能关系为双六边形锥体>八面体形>纺锤体形。可见,采用不同调节剂对所合成MOFs的形貌和应用性能均产生影响。吸附是界面化学反应的重要基础过程,将MOFs作为吸附剂的研究已有较多报道[21];然而,关于采用不同调节剂合成MOFs的性质和吸附性能的研究却少见。因此,研究不同调节剂合成MOFs的性质及其吸附性能的差异,可为揭示不同调节剂合成MOFs的应用性能差异提供基础资料,也可为MOFs的性能优化及化学反应过程的调控提供必要的指导。
MIL-100(Fe)是一种被广泛研究的 MOFs材料[22-25],以HF溶液为调节剂是合成MIL-100(Fe)最常用的方法[26]。由于HF的强腐蚀作用,以HF溶液为调节剂合成的MIL-100(Fe)不适合用玻璃容器作反应场所,且由于F-的配位性强而不利于提高样品的吸附性能。因此,有必要研究用其他调节剂替代HF合成MIL-100(Fe)。根据文献[18-20,26-27],无机酸、H2O、NaAc等酸碱性不同的试剂均可作为合成MOFs的调节剂。然而,迄今未见以HCl、H2O和NaAc为调节剂合成MIL-100(Fe)样品,进而研究其微观结构和表面性质的报道。因此,本文选用HF、HCl、H2O和NaAc四种调节剂合成了不同的MOFFe样品,用 X 射线衍射(XRD)、透射电镜(TEM)、N2等温吸附-脱附、综合热分析(TG/DTG和DTA)、质子电位滴定等手段对样品的结构和表面性质进行了表征分析,并研究了样品对Seビ的吸附性能。这些研究有助于发展性能优良的水体Seビ吸附材料,可为水体系中硒的污染治理提供基础资料和实践指导,同时也可为深入理解MOFs表面性质与吸附等应用性能的构效关系提供基础研究资料。
1 实验方法
1.1 实验试剂
所用试剂均为分析纯级,实验用水由超纯水机(HK-UP-11-20,浩康科技)制备。
1.2 样品制备
参照文献[26]的方法合成MOF-Fe样品:取0.015 mol FeCl3·6H2O于聚四氟乙烯容器中,加入75 mL超纯水溶解,再加入0.010 mol均苯三甲酸和0.030 mol HF;将聚四氟乙烯容器放入水热合成反应釜并置于鼓风干燥箱中,在150℃下合成84 h。自然降至室温,将产物离心并用超纯水洗涤3次至上清液无色,再用无水乙醇浸泡4 h并离心,重复2次得到沉淀,在鼓风干燥箱中80℃下烘干沉淀得到MOFFe(HF)样品。将HF依次用等物质的量的HCl、H2O和NaAc替代合成MOF-Fe,样品分别记为MOF-Fe(HCl)、MOF-Fe(H2O)和 MOF-Fe(NaAc)。
1.3 材料表征
样品的XRD分析采用粉末压片法,在X射线衍射仪(XRD-7000型,岛津)上进行,测试条件:Cu Kα辐射 (波长λ=0.154 056 nm),工作电压 40 kV、工作电流 30 mA,步长为 0.01°,扫描速度为 8°·min-1, 扫描范围为 2°~30°。样品的 TEM 测试在JEM-1400型透射电镜仪上进行,加速电压为80 kV。样品的N2等温吸附-脱附在N2等温吸附-脱附分析仪(AutosorbiQstation,Quantachrome Instruments)上测试,样品在150℃下脱气10 h,测试温度为77 K,相对压力(p/p0)范围为10-6~0.994。 用多点 Brunauer-Emmett-Teller(BET)方法计算样品的比表面积,根据p/p0为0.994时的N2吸附体积计算样品的总孔体积,用t-plot方法计算样品的微孔体积,用Non-Local Density Functional Theory(NLDFT)方法计算样品的孔径分布。样品的综合热分析(TG/DTG和DTA)分析在综合热分析仪(SⅡTG/DTA6300,日本精工)上进行,氩气气氛下测试,温度范围为50~800℃,升温速率为10℃·min-1。样品的质子电位滴定分析在自动电位滴定仪(836 Titrando,Metrohm Swiss)上进行,主要参数为:滴定液以每次0.05 mL等份加入,搅拌2 min后测定悬浮液电位,当电位漂移率小于0.5 mV·min-1时继续下次滴定,当2次滴定间隔时间超过20 min后则达到滴定终点。根据改进的Davies方程,得到不同离子强度条件下样品的pH-qH关系曲线,2种不同离子强度下的pH-qH关系曲线交点即为样品的电荷零点(pHPZC)。
1.4 吸附实验
配制0.01 mol·L-1的KCl溶液,并调节pH值至5.0。称取0.90 g MOF-Fe粉末样品置于300 mL上述KCl溶液中,超声分散10 min,在磁力搅拌状态下用 1.0 mol·L-1的 NaOH 溶液和 1.0 mol·L-1的HCl溶液调节悬浮液的pH值至5.0,并稳定24 h,得到3 g·L-1的吸附剂悬浮液。称取0.691 8 g亚硒酸钠,用少量上述KCl溶液溶解,转移并用KCl溶液定容至100 mL容量瓶中得到40 mmol·L-1的Seビ储备液;然后将Seビ储备液用KCl溶液分别稀释5 倍和 20 倍,得到 8 mmol·L-1和 2 mmol·L-1的 Seビ溶液;再用 1.0 mol·L-1的 NaOH 溶液和 1.0 mol·L-1的HCl溶液调节Seビ溶液的pH值至5.0,即为Seビ使用液。
分别取10 mL吸附剂悬浮液于若干离心管中,依次加入不同体积Seビ使用液(2或8 mmol·L-1),最后补加KCl溶液至30 mL,使吸附体系中吸附剂浓度为 1.0 g·L-1,Seビ初始浓度从 0 mg·L-1梯度增大至 395.87 mg·L-1。 在 25℃、转速 250 r·min-1条件下振荡24 h。将振荡后的悬浮液离心,用氢化物发生-原子荧光分光光度计(AFS-8330,北京吉天)测定上清液中Seビ的浓度。根据吸附前后溶液中Seビ的浓度差计算样品对Seビ的吸附量,实验重复3次,取平均值。
2 结果与讨论
2.1 样品的结构与形貌
图1为样品的XRD图。MOF-Fe(HF)样品的主要衍射峰对应的 2θ角度依次为 3.46°、4.08°、4.90°、5.28°、5.90°、6.26°、10.22°、11.00°、12.52°、14.88°、17.76°、18.56°、18.86°、20.06°、24.02°、27.60°和28.00°,与文献[26]报道一致。 MOF-Fe(HCl)、MOF-Fe(H2O)和MOF-Fe(NaAc)的主要衍射峰与MOF-Fe(HF)样品主要衍射峰一致,没有出现杂质衍射峰,但是相应衍射峰的强度有所降低。这表明用HF、HCl、H2O和NaAc作为调节剂合成的4种样品均具有MIL-100(Fe)的晶体结构,但用HCl、H2O和NaAc合成的样品的结晶度略低。根据文献报道[28],晶体材料XRD图中各衍射峰的相对强度变化可反映样品结构中晶面取向的差异。MOF-Fe(HF)的XRD图中3个最强衍射峰的 2θ角度依次为 11.00°、3.46°和4.08°,MOF-Fe(HCl)的依次为 10.94°、4.05°和 3.44°,MOF-Fe(H2O)的依次为 4.10°、11.00°和 3.50°,MOFFe(NaAc)的依次为 10.92°、3.41°和 4.02°。 可见,4 种样品的XRD图中,3个最强衍射峰的相对峰强差异明显。因此,尽管4种样品均具有MIL-100(Fe)的晶体结构,但是其结晶度和晶面取向不同。
图2为样品的TEM图像,4种样品的形貌均为八面体形。MOF-Fe(HF)的典型颗粒粒径为150~300 nm,大颗粒中混杂着小颗粒和少量不规则颗粒。MOF-Fe(HCl)的颗粒粒径较均一,大多约为100 nm。MOF-Fe(H2O)的颗粒粒径集中分布在200~300 nm之间,同时有少量小颗粒物。MOF-Fe(NaAc)的颗粒粒径主要在200~400 nm之间,同时存在较多不规则的细颗粒。

图1 样品的X射线衍射图Fig.1 X-ray diffraction patterns(XRD)of the samples

图2样品的透射电镜(TEM)图片Fig.2 Transmission electron microscope(TEM)of the samples
2.2 比表面积及孔性结构
图3 为样品的N2等温吸附-脱附及孔径分布曲线。4种样品的N2等温吸附-脱附曲线较类似。当相对压力(p/p0)小于0.2时,样品对N2的吸附体积迅速增大;随着p/p0的增大,N2吸附体积缓慢增加;在p/p0>0.9时,N2吸附体积迅速增大并趋于最大值,吸附-脱附曲线形成微弱的滞后环。4种样品的N2等温吸附曲线均符合BDDT分类的Ⅳ型吸附等温线,表明采用不同调节剂合成的样品的孔性结构类似,且均为微孔和介孔混合孔样品。

图3 样品的N2等温吸附-脱附曲线Fig.3 N2adsorption-desorption curves of the samples

表1 样品的孔性结构及比表面积数据Table 1 Pore structural properties of the samples
样品的比表面积及孔性结构数据列于表1。样品的BET比表面积大小关系为MOF-Fe(NaAc)>MOF-Fe(HF)>MOF-Fe(H2O)>MOF-Fe(HCl)。 用 HF、HCl、H2O和NaAc 4种调节剂合成的样品的总孔体积依次减小。4种样品的介孔孔径分布峰均为2.50 nm;MOF-Fe(HCl)和 MOF-Fe(H2O)样 品 在 孔 径 为2.03 nm处的孔体积含量占比较大,而MOF-Fe(HF)和MOF-Fe(NaAc)样品在孔径为2.03 nm处的孔体积含量占比较小;4种样品的微孔孔径分布峰依次为 1.27、1.22、1.22 和 1.17 nm。 根据文献[29],F-和 Cl-的离子半径分别为0.133和0.181 nm,而Ac-半径最大。MOF-Fe(HF)与MOF-Fe(HCl/H2O)样品的微孔孔径峰差值0.50 nm与F-和Cl-的离子半径差值0.48 nm接近,同时F-、Cl-和Ac-三种离子的半径关系与相应样品的微孔孔径峰成负相关。根据文献报道[30],调节剂可以影响晶体材料的晶胞参数,导致晶粒的晶面取向、尺寸/形貌产生相应的变化;MIL-100(Fe)的晶体结构单元是由(μ-O2-)连接的3个铁氧八面体组成[21],样品的XRD衍射峰显示不同样品的晶面取向不同,这表明改变合成MIL-100(Fe)所用的调节剂可对铁氧八面体的晶胞参数产生影响,导致其晶体形貌/尺寸发生变化,进而影响其孔径分布。样品的孔径分布表明,MOF-Fe样品的微孔孔径可能受合成体系中调节剂种类调控,通过向合成MOFFe的体系中加入不同的调节剂可以起到调控MOFFe样品微孔孔径的作用。
2.3 热分析
图4为样品的热重(TG/DTG)曲线。MOF-Fe(HF)样品的TG曲线有3个失重阶段,当温度在50~91℃之间,样品的失重量为11.0%;当温度在91~289℃之间,失重量为7.7%;当温度在289~522℃之间,失重量为45.4%;样品的总失重量为64.1%。MOFFe(HCl)样品的TG曲线中,温度为50~89℃时的失重量为5.1%,温度为89~315℃时的失重量为7.9%,温度为315~499℃时的失重量为44.2%,温度高于499℃时样品持续缓慢失重至趋于稳定,样品的总失重量为63.7%。MOF-Fe(H2O)样品中,温度为 50~92℃、92~310℃和 310~507℃时的失重量分别为5.4%、11.5%和42.8%;在温度为566~716℃时,样品的TG曲线中出现了失重量为17.8%的失重阶段;样品的总失重量为79.1%。MOF-Fe(NaAc)样品的TG曲线与MOF-Fe(H2O)样品类似,其失重阶段温度范围分别为50~109℃(失重量为7.6%)、109~297 ℃(失重量为 7.2%)、297~507 ℃(失重量为46.6%)和 563~703 ℃(失重量为 7.0%),样品的总失重量为72.1%。
图5为样品的差热分析(DTA)曲线。在温度较低(≤109℃)时,样品出现了一个微弱的吸热峰;当温度继续升高至525℃时,样品的DTA曲线中先后出现了弱吸热峰和强放热峰;当温度继续上升时,在MOF-Fe(HCl)、MOF-Fe(H2O)和 MOF-Fe(NaAc)样品的DTA曲线中出现了较明显的宽且弱的吸热峰。

图4 样品的TG/DTG曲线Fig.4 TG/DTG curves of the samples
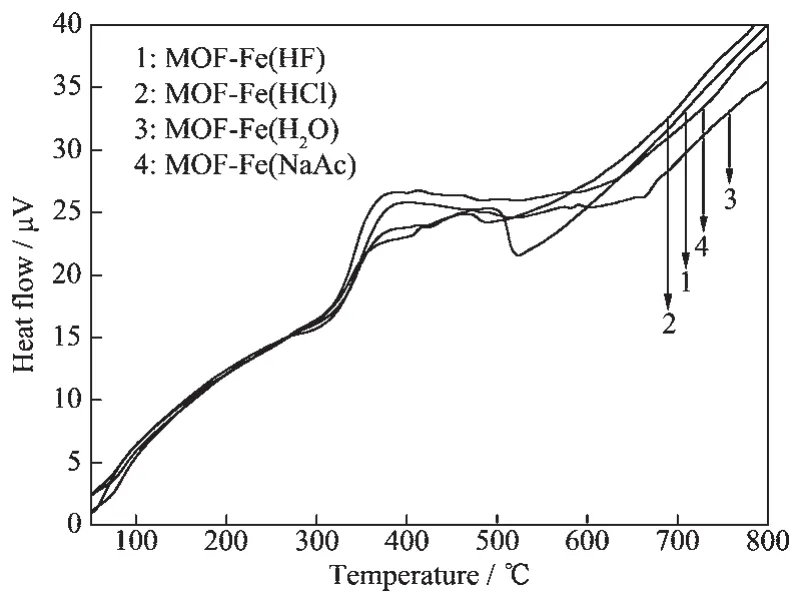
图5 样品的差热(DTA)曲线Fig.5 DTA curves of the samples
根据样品的TG/DTG和DTA曲线可知,当温度≤109℃时,样品因吸附水损失而形成失重阶段。当温度为108~288℃时,样品因失去客体分子而形成失重阶段。当温度为288~522℃时,4个样品DTG曲线中均先后出现1个肩峰和2个明显的失重峰,相应的失重峰依次为脱羟基化反应(2OH-→O2-+H2O)[30]、有机质组分脱羧基[31]和苯环碳化[31]所致;图 4 显示 ,MOF-Fe(HF)、MOF-Fe(H2O)、MOF-Fe(HCl)和MOF-Fe(NaAc)样品的脱羧基失重峰温度分别为415、410、411和410℃,样品的苯环碳化失重峰温度分别为462、467、468和468℃。在MOF-Fe(H2O)和MOF-Fe(NaAc)样品中,当温度分别为566~716℃和563~703℃时,样品热分解形成的氧化铁被碳还原产生失重阶段[32];尽管MOF-Fe(HCl)样品的TG曲线中也存在氧化铁被碳还原而产生的持续失重阶段,但对应的温度范围界限不清晰。
与 MOF-Fe(H2O)、MOF-Fe(HCl) 和 MOF-Fe(NaAc)样品相比,MOF-Fe(HF)样品的脱羧基化温度最高而苯环碳化温度最低,这说明合成MOF-Fe样品时,HF调节剂可增强样品中羧基与苯环之间的C-C键合强度和降低苯环碳化温度。在相对高温区,用HF作为调节剂合成的MOF-Fe样品中没有出现氧化铁被碳还原的失重平台,这说明用HF合成的MOF-Fe样品受热后形成氧化铁的稳定性增强。
2.4 表面电荷
图6为样品的质子电荷量(qH)与pH的关系曲线。4种样品表面均具有可变电荷,在离子强度分别为0.01和0.05 mol·L-1KCl介质中,当悬浮液pH高于电荷零点(pHZPC)时,MOF-Fe(HCl)和 MOF-Fe(H2O)样品质子滴定过程中qH随pH的变化程度明显大于MOF-Fe(HF)和MOF-Fe(NaAc)。这表明在相对碱性条件(pH>pHZPC)下,MOF-Fe(HCl)和 MOF-Fe(H2O)样品中羟基密度高于MOF-Fe(HF)和MOF-Fe(NaAc)。当悬浮液pH小于pHZPC时,MOF-Fe(HF)和MOF-Fe(NaAc)的qH随pH呈近似线性变化,而MOF-Fe(HCl)和MOF-Fe(H2O)的qH随pH的降低而急剧升高。可见,MOF-Fe(HF)和MOF-Fe(NaAc)样品中结构羟基和配位水分子的密度低于MOF-Fe(HCl)和MOF-Fe(H2O)。样品的质子电荷滴定结果列于表2。4种样品的pHZPC关系为MOF-Fe(HF)>MOF-Fe(HCl)=MOF-Fe(NaAc)>MOF-Fe(H2O)。在 0.01 mol·L-1KCl介质中,4种样品悬浮液pH值从6.0降低至3.0时消耗的 H+量分别为 0.125、0.142、0.172 和 0.125 mmol·g-1,这表明4种样品中羟基和配位水分子的密度大小关系为 MOF-Fe(H2O)>MOF-Fe(HCl)>MOFFe(HF)=MOF-Fe(NaAc)。
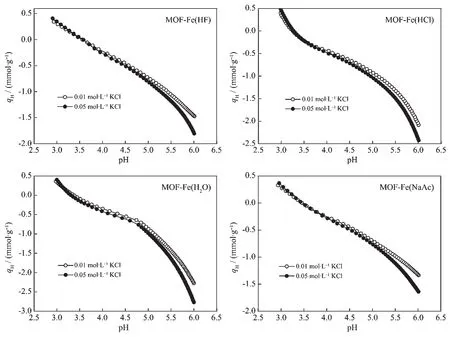
图6 样品的qH-pH关系曲线Fig.6 Proton titration curves of the samples as a function of pH

表2 样品的质子电位滴定结果Table 2 Results of proton potentiometric titrations of the samplesa
MOF-Fe样品中的铁羟基属于 Brønsted碱位点,样品失去配位水分子后的配位不饱和Fe原子成为Lewis酸位点,而Brønsted碱位点和Lewis酸位点通常均为固相材料界面吸附/催化等功能的酸碱活性位点[33]。样品的质子滴定数据显示,用H2O作为调节剂合成的MOF-Fe样品具有最大的Brønsted碱位点和 Lewis酸位点密度,而 HF和NaAc为调节剂合成的样品中相应的酸碱活性位点密度较小。与Cl-相比,F-具有更强的电负性,与Fe原子有较强的相互作用,较难被铁羟基等其他活性阴离子取代[34],而乙酸根中的2个O原子可以与Fe原子以共价键和配位键形式而形成稳定的结构。因此,用HF和NaAc作为调节剂均不利于提高MOFFe样品中活性羟基和配位不饱和Fe原子的密度,导致MOF-Fe样品中的酸碱活性位点的密度较小。
2.5 样品对Seビ的吸附
图7为样品对Seビ的等温吸附曲线。初始pH值为5.0时,吸附体系中Seビ的初始浓度从0升高至395.87 mg·L-1的过程中,4种样品对Seビ的吸附量(Q)均处于增大的趋势。当Seビ初始浓度相同时,4种样品对Seビ的吸附量大小关系为MOF-Fe(H2O)>MOF-Fe(HCl)>MOF-Fe(NaAc)>MOF-Fe(HF)。 当Seビ的初始浓度为 395.87 mg·L-1时,MOF-Fe(H2O)、MOF-Fe(HCl)、MOF-Fe(NaAc)和 MOF-Fe(HF)样品对Seビ的最大吸附量分别为 88.89、84.20、64.07 和60.38 mg·g-1。
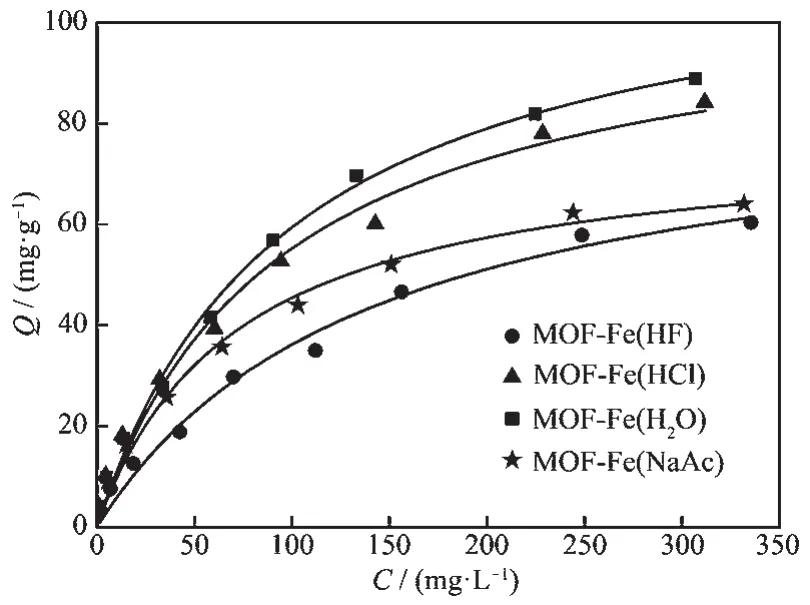
图7 样品对Seビ的等温吸附曲线Fig.7 Adsorption isotherms of Seビby the samples
用Freundlich模型和Langmuir模型对样品吸附Seビ的等温吸附数据拟合,Freundlich模型和Langmuir模型表达式如下:

式中:Q表示吸附剂对吸附质的吸附量 (mg·g-1),C为吸附体系中吸附质的平衡浓度(mg·L-1)。Freundlich模型中k为与吸附容量相关的常数(mg1-1/n·L1/n·g-1),1/n为与吸附强度相关的经验常数。Langmuir模型中Qm表示吸附剂对吸附质的饱和吸附容量(mg·g-1),b为与吸附结合能及亲和力相关的常数(L·mg-1)。
四种样品吸附Seビ的实验数据模型拟合参数列于表3。Freundlich模型的拟合参数中,参数1/n可反映样品吸附Seビ的吸附强度[35]。MOF-Fe(HF)、MOF-Fe(HCl)、MOF-Fe(H2O)和 MOF-Fe(NaAc)4 种样品的拟合系数 1/n分别为 0.53、0.47、0.49和0.43,这表明样品对Seビ的吸附强度关系为MOFFe(NaAc)>MOF-Fe(HCl)>MOF-Fe(H2O)>MOF-Fe(HF)。Langmuir模型的拟合结果中,MOF-Fe(HF)、MOF-Fe(HCl)、MOF-Fe(H2O)和 MOF-Fe(NaAc)四种样品对 Seビ的饱和吸附容量(Qm)分别为87.15、107.07、117.40和 77.69 mg·g-1, 拟合常数 b 分别为 0.007、0.011、0.010和0.014 L·mg-1,说明4种样品对Seビ的吸附亲和力关系为 MOF-Fe(NaAc)>MOF-Fe(HCl)>MOFFe(H2O)>MOF-Fe(HF),这与 Freund-lich 模型拟合结果一致。2种模型对等温吸附数据的拟合度(R2)显示,4种样品对Seビ的等温吸附过程更适合用单分子层吸附的Langmuir模型描述。

表3 样品吸附Seビ的等温吸附模型拟合参数Table 3 Equilibrium isothermal adsorption parameters of Seビby the samples
固-液吸附体系中,固体样品的表面吸附能力与其表面性质密切相关。为了考察4种MOF-Fe的表面性质对其吸附Seビ的影响,以样品的比表面积(SBET)、总孔体积(Vtotal)、微孔体积(Vmicro)、pH 值由 5.0降低至各样品pHZPC的滴定过程中样品的羟基消耗量和pH=5.0时的质子电位(qH)为参数,以样品吸附Seビ的Qm为因变量,对样品的表面性质参数与吸附Seビ的Qm做显著性分析 (所用软件版本为 Origin 8.0),所得的Pearson相关系数(Pearson coefficient)和显著性水平(P)列于表4。
样品的 SBET、Vtotal、Vmicro、qH等表面性质参数与 Qm之间的P均大于0.05,说明Qm与样品的SBET、Vtotal、Vmicro和qH等表面性质之间没有显著相关性。样品的与Qm之间相关性的p<0.05,相关系数为0.964, 说明Qm与ΔnOH-之间呈现出显著正相关性。MOFs材料对无机含氧阴离子酸根的吸附主要包括阴离子交换、静电作用等吸附机制[21,36];吸附剂吸附污染物时,样品的有效比表面积和有效孔径是决定样品对污染物吸附容量的重要因素。本研究中的显著性分析表明:当吸附体系pH值为5.0时,MOF-Fe与Seビ之间的吸附机理主要为Seビ与羟基之间的离子交换等化学吸附;尽管qH与Qm之间没有显著相关性,但在pH值为5.0的条件下带负电荷的样品与Seビ之间存在静电排斥作用,因此静电作用也会影响表面吸附行为;样品的比表面积、微孔孔体积和总孔体积与样品对Seビ的饱和吸附容量之间没有显著相关性,这可能是4种调节剂合成的MOF-Fe样品中的有效比表面积与有效孔体积均没有明显的差异所致。由样品的表面性质与对Seビ的饱和吸附容量的相关性分析可知,在合成MOF-Fe的体系中,用H2O和HCl作为调节剂合成的样品具有较大的羟基等活性位点密度,而HF和NaAc合成的样品的表面活性位点密度相对较小。因此,可以通过选择合适的调节剂合成MOF-Fe样品,以提高样品的表面活性位点密度和表面吸附能力。

表4 样品的显著性分析结果Table 4 Results of significance analysis about the samples
3 结 论
(1)以 HF、HCl、H2O和 NaAc作为调节剂合成的MOF-Fe均具有八面体MIL-100(Fe)晶体结构,然而4种样品表面性质差异明显。4种样品的BET比表面积为 1 517~1 734 m2·g-1,样品的总孔体积和微孔孔径受调节剂控制。用HF为调节剂可提高样品的脱羧基温度,降低苯环碳化温度;用HCl、H2O和NaAc为调节剂可促使样品热分解形成的铁氧化物与碳之间发生氧化还原反应。MOF-Fe(H2O)样品的Brnsted碱位点和Lewis酸位点等活性位点密度最大,而MOF-Fe(HF)和MOF-Fe(NaAc)样品中相应的酸碱活性位点较小。
(2)初始pH值为5.0时,4种样品对Seビ的等温吸附过程均更适合用Langmuir模型描述,样品对Seビ的饱和吸附容量大小关系为MOF-Fe(H2O)>MOF-Fe(HCl)>MOF-Fe(HF)>MOF-Fe(NaAc),对 Seビ的吸附亲和力关系为MOF-Fe(NaAc)>MOF-Fe(HCl)>MOF-Fe(H2O)>MOF-Fe(HF)。 样品中羟基密度与样品对Seビ的吸附吸附容量呈显著正相关;这表明,通过选择合成MOF-Fe所用的调节剂可以有效改变样品中羟基等活性位点密度,进而改善样品对Seビ的饱和吸附容量等吸附性能。
参考文献:
[1]Ju W,Li W,Li Z,et al.J.Trace Elem.Med.Biol.,2017,44:8-16
[2]Winkel L H,Johnson C A,Lenz M,et al.Environ.Sci.Technol.,2011,46(2):571-579
[3]Fernández-Martínez A,Charlet L.Rev.Environ.Sci.Biotechnol.,2009,8(1):81-110
[4]GB 3838-2002,Environmental Quality Standards for Surface Water(地表水环境质量标准).2002.
[5]ZHU Jian-Ming(朱建明),QIN Hai-Bo(秦海波),LI Lu(李璐),et al.Acta Scientiae Circumstantiae(环境科学学报),2008,28(4):772-777
[6]Santos S,Ungureanu G,Boaventura R,et al.Sci.Total Environ.,2015,521-522:246-260
[7]WANG Lin(王琳),SHI Yong-Sheng(施永生).Treatment of Waster Water Contain Selenium(含硒水处理).Beijing:Chemistry Industry Press,2005.
[8]Hu B W,Ye F,Jin C G,et al.Chemosphere,2017,184:408-416
[9]Xia X F,Ling L,Zhang W X.Chemosphere,2017,168:1597-1603
[10]Chan Y T,Kuan W H,Chen T Y,et al.Water Res.,2009,43(17):4412-4420
[11]Ma Z Y,Shan C,Liang J L,et al.Chemosphere,2018,193:134-141
[12]Rosi N L,Eckert J,Eddaoudi M,et al.Science,2003,300(5622):1127-1129
[13]Sun J K,Xu Q.Energy Environ.Sci.,2014,7(7):2071-2100
[14]Alezi D,Belmabkhout Y,Suyetin M,et al.J.Am.Chem.Soc.,2015,137(41):13308-13318
[15]Cao X H,Zheng B,Shi W H,et al.Adv.Mater.,2015,27(32):4695-4701
[16]Chen Q S,Sun J L,Li P,et al.J.Am.Chem.Soc.,2016,138(43):14242-14245
[17]HUANG Gang(黄刚),CHEN Yu-Zhen(陈玉贞),JIANG Hai-Long(江海龙).Acta Chim.Sinica(化学学报),2016,74(02):113-129
[18]Guo H L,Zhu Y Z,Wang S,et al.Chem.Mater.,2012,24(3):444-450
[19]Cheng X Q,Zhang A F,Hou K K,et al.Dalton Trans.,2013,42:13698-13705
[20]Liu Y L,Gao P F,Huang C Z,et al.Sci.China Ser.B:Chem.,2015,58(10):1553-1560
[21]Kumar P,Pournara A,Kim K H,et al.Prog.Mater.Sci.,2017,86:25-74
[22]Patra S,Sene S,Mousty C,et al.ACS Appl.Mater.Interfaces,2016,8(31):20012-20022
[23]Bghadra B N,Jhung S H.ACS Appl.Mater.Interfaces,2016,8(10):6770-6777
[24]Zhu N N,Liu X H,Li T,et al.Inorg.Chem.,2017,56(5):3414-3420
[25]Liu X C,Zhou Y Y,Zhang J C,et al.ACS Appl.Mater.Interfaces,2017,9(24):20255-20275
[26]Song G Q,Wang Z Q,Wang L,et al.Chin.J.Catal.,2014,35(2):185-195
[27]Lee Y R,Kim J,Ahn W S.Korean J.Chem.Eng.,2013,30(9):1667-1680
[28]ZHANG Su(张素),WANG Hai-Shui(王海水).Chinese J.Inorg.Chem.(无机化学学报),2013,29(1):31-35
[29]ZHAO Xin-Hua(赵新华),CHEN Lin(陈玲),CAO Yong-Ge(曹永革),et al.Foundation of Solid-State Inorganic Chemistry and the Design Synthesis of Novel Materials(固体无机化学基础及新材料的设计合成).Beijing:Higher Education Press,2012:128-131,610-611
[30]Cornell R M,Schwertmann U.The Iron Oxides,Structure,Properties,Reactions Occurences,Uses.Weinheim:Wiley-VCH,2003.
[31]Zhang L.Thesis for the Doctorate of Michigan Technological University.2011.
[32]JIANG Wu-Feng(蒋 武 锋),ZHAO Shuo(赵 朔),HAO Su-Ju(郝素菊),et al.Chinese Journal of Process Engineering(过程工程学报),2017,17(2):362-366
[33]Hattori H,Ono Y,Translated by GAO Zi(高滋),LE Ying-Hong(乐英红),HUA Wei-Ming(华伟明).Solid Acid Catalysis(固体酸催化).Shanghai:Fudan University Press,2016.
[34]Kuang L Y,Liu Y Y,Fu D D,et al.J.Colloid Interface Sci.,2017,490:259-269
[35]Hajjaji W,Ganiyu S O,Tobaldi D M,et al.Appl.Clay Sci.,2013,83-84:91-98
[36]Chen L,Ji T,Mu L W,et al.J.Environ.Manage.,2017,196:168-177
猜你喜欢
中成药(2018年2期)2018-05-09
现代园艺(2018年3期)2018-02-10
新乡学院学报(2016年6期)2016-12-01
中国塑料(2016年12期)2016-06-15
中国人兽共患病学报(2016年6期)2016-01-30
人间(2015年11期)2016-01-09
中国继续医学教育(2015年5期)2016-01-07
中国塑料(2015年3期)2015-11-27
制造技术与机床(2015年3期)2015-01-27
中国塑料(2014年2期)2014-10-17
