
水溶性膦酸双核金属钌催化t-BuOOH氧化醇性能
2018-05-05 06:22廖海深易小艺
无机化学学报 2018年5期
廖海深 李 辰 易小艺*,,2
(1中南大学化学化工学院,长沙 410083)
(2锰资源高效清洁利用湖南省重点实验室,长沙 410083)
将醇氧化成醛或者酮的反应,在精细化工、制药等方面有不可忽视的地位,科学家一直致力实现经济、绿色氧化的目标,如使用廉价、可循环使用的催化剂,环境友好的O2,H2O2作为氧化剂,而在温和条件,水溶液体系中完成这一氧化过程也是追求目标之一。用水溶液作为溶剂体系,不仅清洁无污染,廉价易得,而且不易挥发和燃烧,安全可靠,对醇氧化反应的工业生成有重要意义。目前,水溶液中醇氧化的研究主要集中在多酸配合物体系,主要原因是多酸具有良好的水溶性[1-2]。因此,寻找水溶性金属配合物往往是研究水溶液中醇氧化的前提。
普通的金属配合物,含有较多的有机基团,水溶性较差。我们的研究中发现,羟基亚乙基二膦酸(hedp)稳定的含钌金属-金属键的配合聚合物(NH4)3[Ru2(hedp)2]·2H2O(图 1)具有层状结构,但有较好的水溶性。该配合物的结构研究表明Hedp膦酸基团和酰胺、羧酸配体一样,螯合2个钌原子形成浆轮型结构单元,2个钌原子分别为+2和+3价,通过RuⅢ-RuⅡ金属-金属键相连[3]。研究表明,氧、氮作为端位配位原子可以与钌原子键合形成Ru=Ru X(X=O,N)电子共轭体系,从而有利于获得较稳定的高氧化态金属中心[4-5]。利用这一性质,这类配合物常常应用到有机底物的催化氧化反应。如Ren Tong课题组发现酰胺配体双核钌配合物[Ru2(HNOCC(CH3)2)4Cl]和[Ru2(HNOCCH2CH3)4Cl],羧酸配体双核钌配合物[Ru2(esp)2Cl](esp=α,α,α′,α′-tetramethyl-1,3-benzenedipropionate),Ru2(OOCCH3)4Cl和膦酸配体双核钌Na4[Ru2(hedp)2(H2O)Cl]能催化 t-BuOOH、H2O2氧化硫醚[6-8];Doyle课题组发现己内酰胺双核铑配合物[Rh2(cap)4]可以催化t-BuOOH的烯丙位氧化[9]。最近,本课题组研究发现hedp双核钌单分子化合物K3[Ru2(hedp)2·2H2O]·6H2O 作催化剂,在(NH4)2Ce(NO3)6或NaIO4作氧化剂条件下,可以催化水氧化生成氧气,通过循环伏安、紫外、质谱等方法可以证实RuⅢ-RuⅣ=O,RuⅢ-RuⅤ=O,RuⅢ-RuⅢOOH 等中间体存在,而这些中间体往往可以和有机底物发生氧化反应。因此,基于以上这些文献调研,让我们很自然想到,可以利用水溶性的膦酸配体双核钌(NH4)3[Ru2(hedp)2]·2H2O作为催化剂实现在水溶液中的催化醇氧化反应。
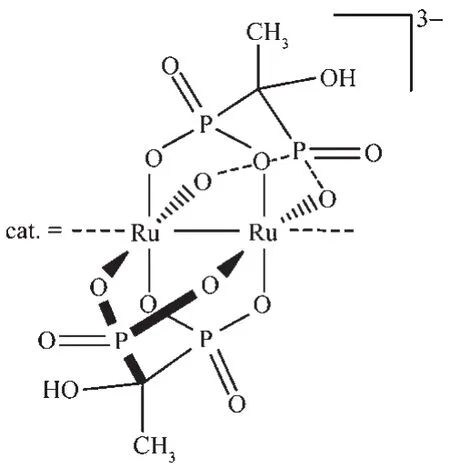
图1 (NH4)3[Ru2(hedp)2]·2H2O的构建单元[Ru2(hedp)2]3-结构图Fig.1 Structure of building block[Ru2(hedp)2]3-in(NH4)3[Ru2(hedp)2]·2H2O
1 实验部分
1.1 仪器和试剂
紫外光谱用国产上海棱光UV759S紫外-可见光谱仪测定;气相色谱用国产南京科捷GC5890气相色谱仪测定。
气相色谱工作条件:色谱柱:SE-30,规格:0.32 mm×30 mm×0.25 μm; 进样温度 250℃, 检测温度300℃(氢火焰),柱箱温度110~160℃(根据产物不同调整)。氢气柱前压0.15 MPa,空气柱前压0.15 MPa,氮气柱前压 0.30 MPa。
配合物(NH4)3[Ru2(hedp)2]·2H2O[3],Na4[Ru2(hedp)2(H2O)Cl][10],K3[Ru2(hedp)2·2H2O]·6H2O[10]按照参考文献的方法合成,其他试剂在百灵威购买。
配合物水溶液配置:称取(NH4)3[Ru2(hedp)2]·2H2O(695 mg,1.0 mmol)置于 90 mL 水溶液中,然后回流5 h,固体完全溶解,溶液冷却至室温,过滤,得到棕红色的澄清水溶液,然后定容至100 mL,获得0.01 mol·L-1配合物水溶液备用。注意:配合物浓度不宜超过0.02 mol·L-1,否则长时间放置,将有溶质固体析出。
1.2 催化反应实验步骤
量取0.01 mol·L-1催化剂水溶液0.5 mL,加水稀释至2 mL,按比例依次加入有机底物(0.5 mmol)、氧化剂(1.5 mmol)。在室温下反应,观测溶液的颜色及状态变化。一定时间后,加入内标碘苯,搅拌均匀,然后取混合溶液100 μL置于1 mL二氯甲烷,加入无水硫酸钠干燥除尽水分,再取液5 μL注入气相色谱仪。分析结果使用内标法计算溶液中底物及产物的含量,底物转化率,产物产率。
2 结果与讨论
2.1 催化剂种类及氧化剂筛选
首先,研究了不同膦酸配体双核钌催化剂和氧化剂种类对苯甲醇氧化的影响。实验条件为:0.5 mmol的苯甲醇作底物,催化剂量为1%(n/n),溶剂2 mL水,室温反应1 h。氧化剂选择为30%的H2O2溶液、70%的 t-BuOOH 水溶液、N-氧化吡啶、4,4′-联氧化吡啶及碘酸钾,用量为1.5 mmol。
实验数据总结于表1。从表1可看出,在含有金属催化剂条件下,H2O2,氧化吡啶,碘酸钾作为氧化剂,苯甲醇转化率不足3%;无金属催化剂,仅有t-BuOOH作为氧化剂时,苯甲醇转化率达8%;而加入金属催化剂,以t-BuOOH作为氧化剂时,苯甲醇转化率明显提高。以 Na4[Ru2(hedp)2(H2O)Cl],K3[Ru2(hedp)2·2H2O]·6H2O作为催化剂,苯甲醇转化率不足50%,而以(NH4)3[Ru2(hedp)2]·2H2O作为催化剂,苯甲醇转化率达82%。因此,选择本研究选择(NH4)3[Ru2(hedp)2]·2H2O作为催化剂,70%的t-BuOOH水溶液作为氧化剂。
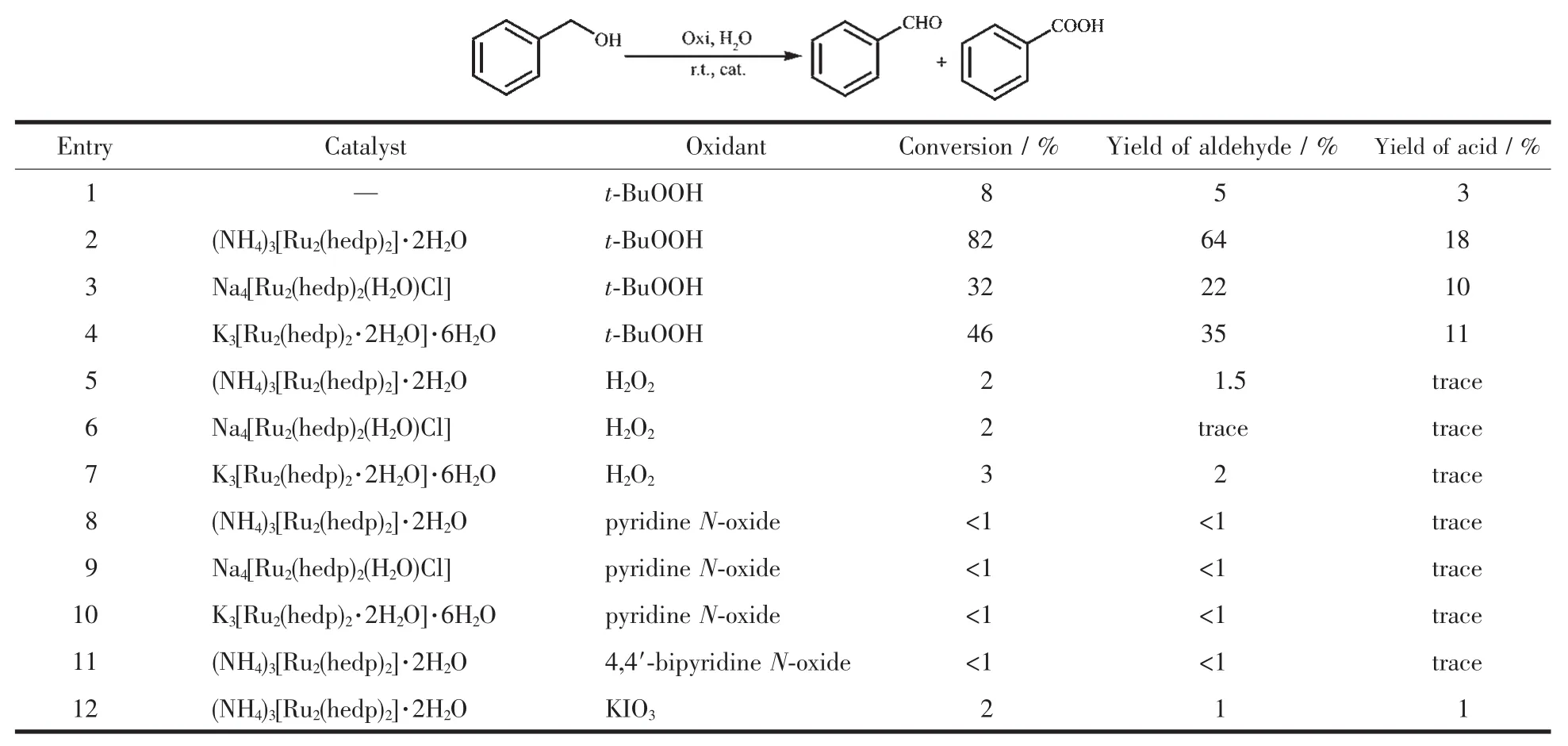
表1 氧化剂种类筛选aTable 1 Oxidant optimization
2.2 催化条件优化
通过实验优化催化条件,如催化剂量,反应时间,氧化剂和底物用量等因素对醇转化率以及产物醛/酸选择性的影响,其实验数据总结于表2。从该表可看出,苯甲醇的转化率随着催化剂用量的增大而增大,催化剂用量由0增至 1%(n/n)时,苯甲醇转化率由8%迅速提升至82%。催化剂用量继续增加到4%(n/n)时,苯甲醇的转化率提升91%,提升幅度有限,同时,反应选择性由1%(n/n)时最高的3.6降至2.8。综合考虑,本研究选择1%(n/n)(NH4)3[Ru2(hedp)2]·2H2O作为催化剂加入量。
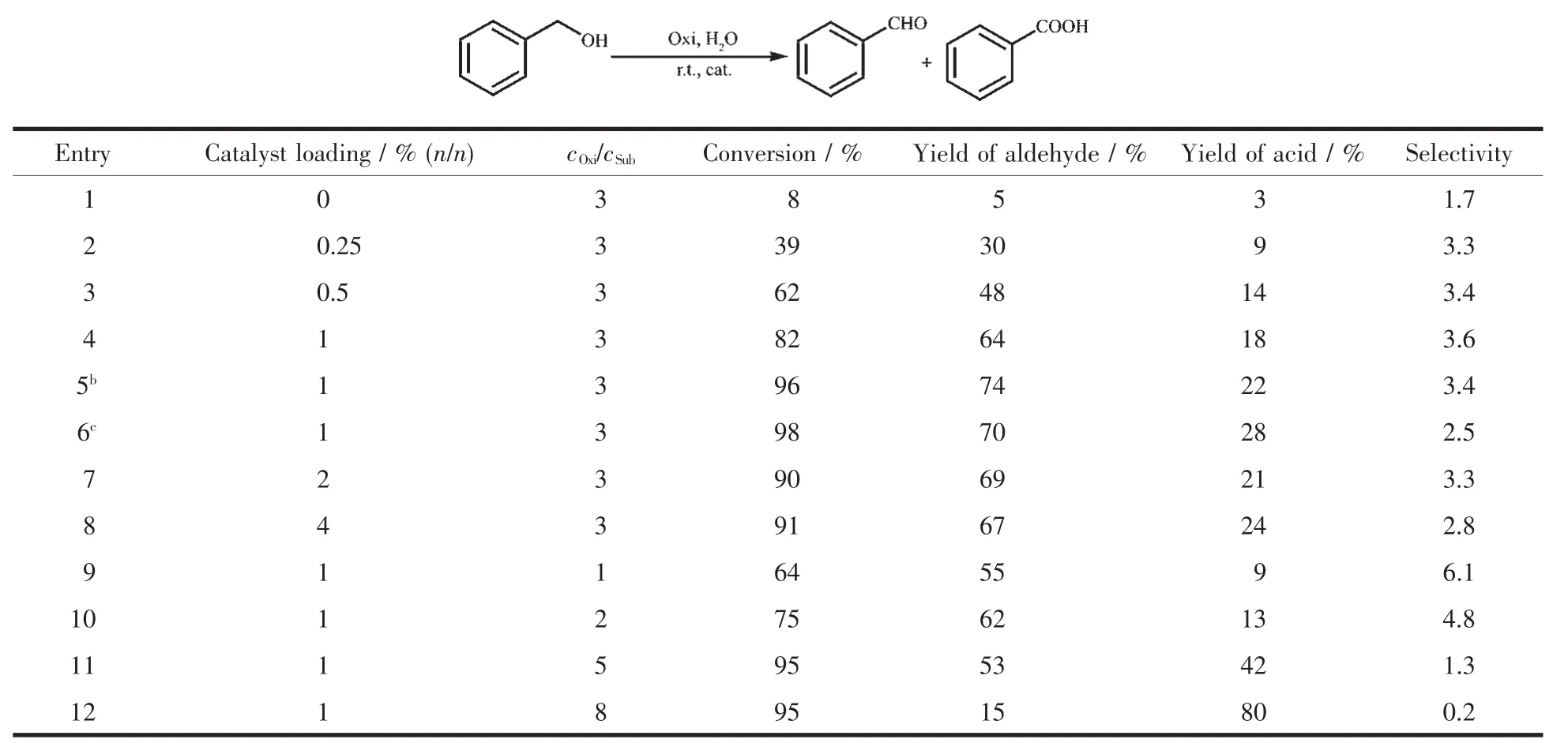
表2 催化条件优化aTable 2 Catalytic condition optimization
从序号4~6的数据可看出,随着时间的延长,(NH4)3[Ru2(hedp)2]·2H2O一直能保持其催化活性,3 h反应时间,几乎可以将苯甲醇完成氧化。从这可得知,催化剂在水溶液中稳定,能保持高活性。
氧化剂相对于底物的用量明显影响苯甲醇转化率和反应选择性(序号 9~12)。当 cOxi/cSub<3 时,苯甲醇的转化率及苯甲醛、苯甲酸的产率都随着氧化剂含量的增加而提高;而后再随着氧化剂的增加,苯甲醛产率下降,苯甲酸产率上升。选择性由cOxi/cSub=3时的3.6下降到cOxi/cSub=8时的0.2。这说明,膦酸双核钌和t-BuOOH生成的中间体,不仅可以氧化苯甲醇,而且可以氧化苯甲醛生成苯甲酸。综合考虑转化率和选择性以及便于比较研究,本催化最优条件确定为0.5 mmol有机底物,1.5 mmol t-BuOOH,1%(n/n)催化剂,水溶液2 mL,室温反应1 h。
2.3 添加物对催化反应影响
表3总结了添加物对催化反应的影响。从该表可知,当加入强或弱的质子酸时,苯甲醇转化率明显下降,选择性提高,而添加碱时,苯甲醇转化率和选择性均有提高。这一现象可以解释为:在催化过程中,涉及质子转移过程,加入酸或碱,分别抑制或有助于质子转移,从而减慢或加快催化反应。另外,当加入碱NH3·H2O时,转化率也有所下降,可能原因是,双核钌和可配位碱NH3和氧化剂t-BuOOH竞争反应,分别形成Ru-NH3配位键和Ru2-OOtBu中间体,从而减慢了催化进程。
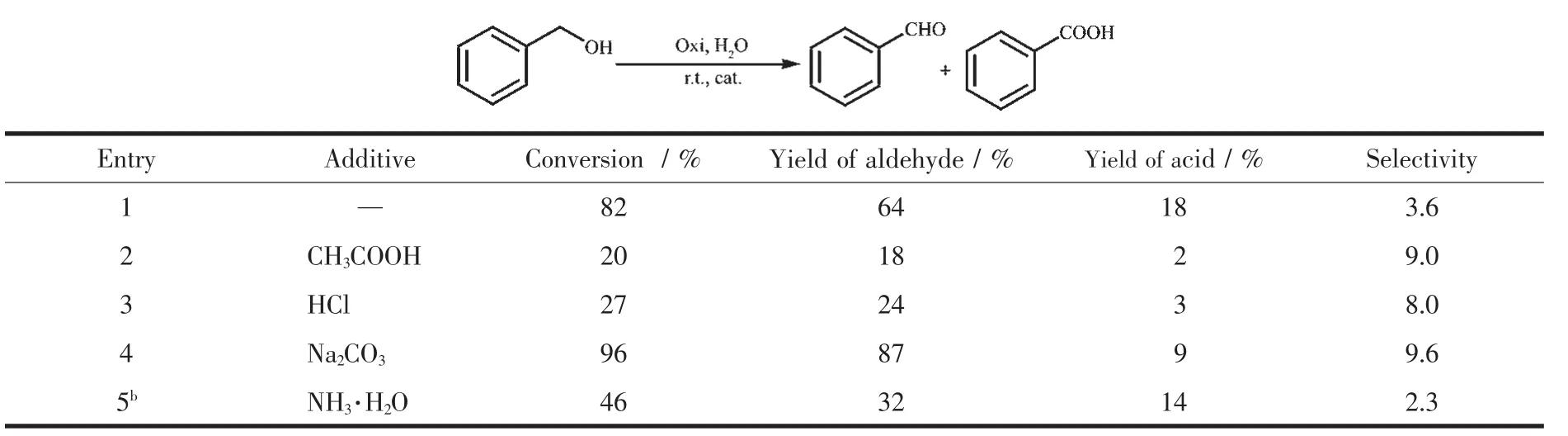
表3 添加物对催化反应的影响aTable 3 Addition of the additive in the catalytic reaction
2.4 醇类底物拓展
本文选择较难氧化的长链醇作为研究对象,考察(NH4)3[Ru2(hedp)2]·2H2O的氧化性能,其数据总结于表4。在1 h内,相对于苯甲醇,长链醇的转化率明显偏低,不足10%,可能的原因是这些长链醇在水溶液中溶解度有限,限制催化反应进行。当反应时间延长至9 h,长链醇几乎完全转化,正辛醇和异辛醇转化率>97%,生成对应的醛和酸,产率分别为72%和20%。而仲辛醇则完全氧化为2-辛酮。
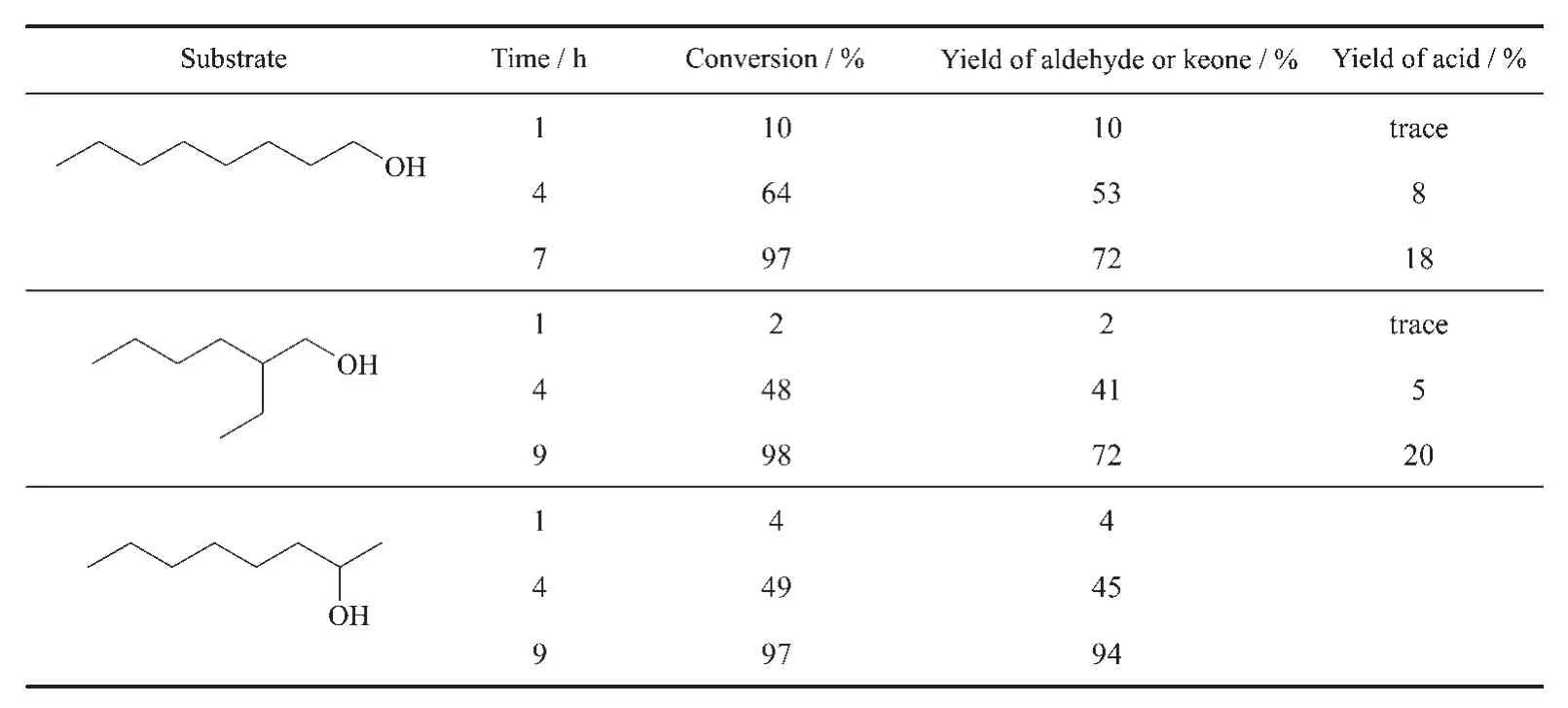
表4 底物拓展Table 4 Oxidation of secondary alcohol and long-chain aliphatic alcohol by(NH4)3[Ru2(hedp)2]·2H2O
2.5 催化机理研究
对于金属配合物催化过氧化合物氧化醇的反应,通常可以根据金属离子和氧化剂形成氧-金属(oxometal pathway)或过氧-金属(peroxometal pathway)活性种的不同而分成两类催化机理[11]。基于以上数据,(NH4)3[Ru2(hedp)2]·2H2O可能经历了过氧金属的反应历程,如图2所示。配合物的双核钌[RuⅢ-RuⅡ]3-单元首先与t-BuOOH反应,形成含过氧键的RuⅢ-RuⅢ-OOtBu中间产物,然后该过氧金属中间体氧化醇生成醛以及氧化醛生成酸。以下几个证据可以说明中间产物RuⅢ-RuⅢ-OOtBu的生成:(1)通过紫外-可见光谱跟踪(NH4)3[Ru2(hedp)2]·2H2O和t-BuOOH(物质的量之比1∶800)的反应,如图3所示,发现(NH4)3[Ru2(hedp)2]·2H2O在456 nm处吸收峰降低,而在329和508 nm处出现新的吸收峰,在412和475 nm处出现等电位点,可以说明,(NH4)3[Ru2(hedp)2]·2H2O和过量t-BuOOH反应,生成了单一反应产物,即Ru2-OOtBu中间产物。反应过程中,颜色明显由棕黄色变到紫色。类似的反应现象和紫外-可见光谱变化也出现在Na4[Ru2(hedp)2(H2O)Cl]和H2O2的反应中,该反应认为有类似的RuⅢ-RuⅢ-OOH中间体产生[8]。 (2)从电化学研究可知,K3[Ru2(hedp)2·2H2O]·6H2O的氧化电位分别为0.68、1.30和1.37 V,能被CeⅣ氧化为 RuⅢ-RuⅢ,RuⅢ-RuⅣ=O 或 RuⅢ-RuⅤ=O,其钌端氧化合物可直接氧化醇生产醛,RuⅢ-RuⅤ=O可以氧化水生成O2。然而,仔细比对(NH4)3[Ru2(hedp)2]·2H2O和t-BuOOH反应获得产物和已知结构的RuⅢ-RuⅢ化合物(H2pip)[(hedp)2(H2O)RuⅢRuⅢ(OH2)](pip=piperazine)[12]和 K3[Ru2(hedp)2·2H2O]·6H2O 和 1 份CeⅣ生成产物的紫外吸收光谱 (三者吸收峰分别为329和 508 nm,321和 505 nm 和 336和 506 nm),发现这三者的吸收峰类似,仅相差几个波数,而明显不同于 K3[Ru2(hedp)2·2H2O]·6H2O 和 CeⅣ(>2 份)生成的RuⅢ-RuⅣ和RuⅢ-RuⅤ产物的紫外吸收光谱,初步推断(NH4)3[Ru2(hedp)2]·2H2O和t-BuOOH反应仅获得单一的 RuⅢ-RuⅢ产物。 (NH4)3[Ru2(hedp)2]·2H2O 和过量的t-BuOOH反应,没有检测到O2生成,基本可以排除 t-BuOOH 氧化[Ru2(hedp)2]3-生成 RuⅢ-RuⅣ和RuⅢ-RuⅤ高氧化态钌端氧中间体的可能,从而可以直接排除高氧化态金属端氧作为中间体直接氧化醇的催化途径。(3)从可能反应机理图2可看出,t-BuOOH氧化(NH4)3[Ru2(hedp)2]·2H2O以及催化过程中,t-BuOOH 和 RuⅢ-RuⅢ生成 RuⅢ-RuⅢ-OOtBu 中间产物过程中伴随质子释放,这一过程也得到实验的印证,即加入酸能抑制Ru2-OOtBu的生成,从而减缓催化反应,而碱的加入,效果刚好相反,但酸对哪一个过程中的RuⅢ-RuⅢ-OOtBu形成有抑制作用不能确定。
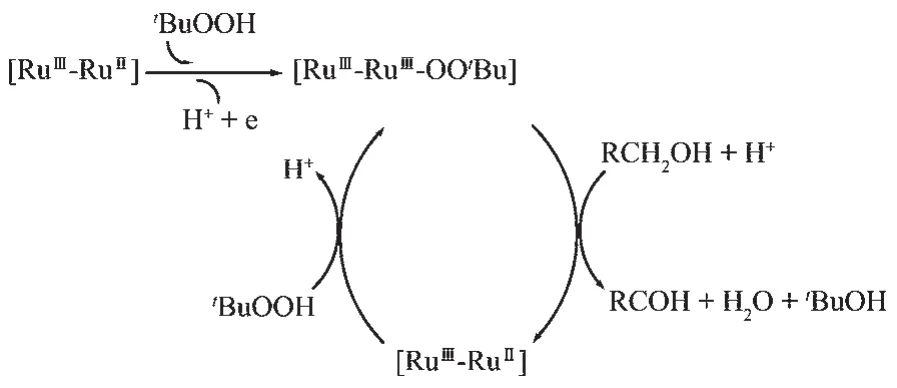
图2 (NH4)3[Ru2(hedp)2]·2H2O催化t-BuOOH氧化醇可能机理Fig.2 Proposed catalytic mechanism of alcohol oxidation with t-BuOOH by(NH4)3[Ru2(hedp)2]·2H2O

图 3 (NH4)3[Ru2(hedp)2]·2H2O 和t-BuOOH(物质的量比1∶800)反应的紫外光谱随时间变化图Fig.3 UV-Vis spectral change with time in molar ratio of(NH4)3[Ru2(hedp)2]·2H2O and t-BuOOH(1∶800,n/n)reaction condition
3 结 论
双核金属钌膦酸配合物(NH4)3[Ru2(hedp)2]·2H2O在常温水溶性体系中,可以高效催化t-BuOOH氧化各种醇类,包括一级醇、二级醇,长链醇,生成相对应的醛(酮)和酸。当nt-BuOOH/nSub<3时,醇氧化主要产物是醛,而当nt-BuOOH/nSub>5时,醇氧化主要产物是酸。酸性水溶性体系抑制(NH4)3[Ru2(hedp)2]·2H2O催化t-BuOOH醇氧化反应,而碱性溶液加速催化反应。催化机理研究表明,双核金属钌膦酸配合物的[RuⅢ-RuⅡ]3-单元与t-BuOOH作用,可以形成含过氧键的RuⅢ-RuⅢ-OOtBu中间体,然后再对底物醇进行氧化。
参考文献:
[1]Sloboda-Rozner D,Alsters P L,Neumann R.J.Am.Chem.Soc.,2003,125:5280-5281
[2]Sheldon R A.Catal.Today,2015,247:4-13
[3]Yi X Y,Zheng L M,Xu W,et al.Inorg.Chem.,2003,42:2827-2829
[4]Pap J S,George S D B,Berry J F,et al.Angew.Chem.Int.Ed.,2008,47:10102-10105
[5]Goberna-Ferrón S,Pea B,Soriano-López J,et al.J.Catal.,2014,315:25-32
[6]Barker J E,Ren T.Inorg.Chem.,2008,47:2264-2266
[7]Villalobos L,Paredes J E B,Cao Z,et al.Inorg.Chem.,2013,52:12545-12552
[8]Thompson D J,Paredes J E B,Villalobos L,et al.Inorg.Chim.Acta,2015,424:150-155
[9]Catino A J,Forslund R E,Doyle M P.J.Am.Chem.Soc.,2004,126:13622-13623
[10]Yi X Y,Liu B,Jiménez-Aparicio R,et al.Inorg.Chem.,2005,44:4309-4314
[11]Sheldon R A,Arends I W C E,Dijksman A.Catal.Today,2000,57:157-166
[12]Liu B,Ding T,Hua W J,et al.Dalton Trans.,2013,42:3429-3433
猜你喜欢
中学生数理化(高中版.高考数学)(2022年1期)2022-04-26
房地产导刊(2022年1期)2022-02-28
中学生数理化(高中版.高二数学)(2020年2期)2020-04-21
原子与分子物理学报(2020年5期)2020-03-17
中学化学(2019年4期)2019-08-06
中学化学(2019年4期)2019-08-06
中学化学(2019年2期)2019-07-08
计算机测量与控制(2017年6期)2017-07-01
浙江农业学报(2017年1期)2017-05-17
浙江大学学报(工学版)(2016年9期)2016-06-05
