
Sneddon综合征合并脾肿大及淋巴结肿大:1例报告及文献复习
2018-04-26 06:49
神经病学与神经康复学杂志 2018年1期
上海交通大学医学院附属仁济医院神经内科,上海 200127
Sneddon综 合 征(Sneddon’s syndrome,SS)是一种广泛累及中小动脉的、罕见的神经皮肤综合征,以反复的卒中发作和皮肤网状青斑为主要临床表现,可累及心、肾、眼等多个脏器。Sneddon在1965年首次报道本病。国外对SS的报道较多,而国内报道较少;且合并脾肿大及淋巴结肿大的SS,目前国内未见报道。本文报道1例合并脾肿大及淋巴结肿大的SS患者的诊疗经过,以提高临床上对该病的认识。
1 病例报告
患者,男性,22岁,因“头晕伴视物重影、行走不稳1年半,加重5个月”于2017年3月23日就诊于上海交通大学医学院附属仁济医院神经内科门诊。
患者自2015年9月开始,无明显诱因下出现头晕伴视物重影、行走不稳,表现为走路摇晃、拖步伴脚踩棉花感。于2015年9月22日至当地医院就诊,头颅磁共振成像(magnetic resonance imaging,MRI)平扫+增强扫描显示右侧基底节区异常信号影,T2加权成像呈高信号影,无强化,考虑良性非肿瘤病变,梗死后或脱髓鞘改变可能(图1A)。脑干MRI平扫+增强扫描未见异常。腰椎穿刺脑脊液检查结果:白细胞计数为2.0×106/L,红细胞计数为0×106/L,白蛋白为151 mg/L,寡克隆区带(-)。当地医院考虑“脑干脑炎可能”,给予甲泼尼龙琥珀酸钠冲击及对症支持治疗。出院后,改为醋酸泼尼松片口服减量维持(具体剂量不详),患者行走症状明显好转,基本恢复正常。
本次入院5个月前,患者自觉行走不稳症状加重,于当地中医院就诊,接受中药调理和激素抗炎治疗(中药名称和激素名称以及用药剂量均不详)3个月后,症状呈缓慢的进行性加重,为进一步诊治,于2017年3月23日收入本院。
入院后追问病史,患者于7年前曾有左侧躯体麻木及双眼斜视病史,左侧躯体麻木持续2 d后好转,口服药物(自称包含激素,但具体名称及剂量不详)1年后,双眼斜视症状完全恢复;约5年前,发现躯干、四肢出现网状青斑,以双小腿为著,天气寒冷时更为明显,洗热水澡后可好转,当时未予重视;4年前,有发作性右耳耳鸣及听力下降(具体程度不详)病史,于当地医院治疗20余天后,右耳耳鸣及听力下降明显好转;近2年来,家属反映患者有记忆力减退及智力下降表现。患者否认既往有高血压、糖尿病、心脏病、肾脏病等慢性疾病史,否认吸烟、饮酒及其他不良嗜好,否认类似疾病家族史。幼时智力发育正常,文化程度为大专。
入院后完善体格检查,测体温为36.4 ℃,心率为80次/min,呼吸频率为18次/min,收缩压为150 mm Hg(1 mm Hg=0.133 kPa),舒张压为101 mm Hg;躯干、四肢可见网状青斑,以四肢末端(尤其是双下肢)明显,网状青斑表现为淡红色、蓝色、紫红色的不规则花边状及裂环状,部分破溃(图2A、B、C),压之不褪色。神经系统专科检查:神志清晰,构音含糊;双眼向右视时有复视,右眼外展露白2 mm,左眼活动正常,双侧瞳孔等大等圆,直径为3 mm,双侧瞳孔对光反射(+++);鼻唇沟对称,伸舌居中;颈软,四肢肌张力正常,四肢肌力为5级;双侧针刺觉正常,四肢位置觉、运动觉、振动觉正常;双侧腱反射亢进,双侧踝阵挛(+),双侧Hoffmann征(+),双侧Barbinski征(+),双侧Kerning征(-),左侧指鼻略笨拙,右侧指鼻尚佳,双侧跟膝胫欠稳准,行走不稳,双足间距增宽,直线行走不能,Romberg征(-),饮水试验(-);简易智力状态检查(mini-mental state examination,MMSE)量表评分为26分(大专文化程度)。
实验室检查结果:尿酸为513 μmol/L(参考范围:155~428 μmol/L)、C反应蛋白为63.3 mg/L(参考范围:≤10 mg/L)、红细胞沉降率为25 mm/h(参考范围:0~15 mm/h)、纤维蛋白原为4.96 g/L(参考范围:2~4 g/L)。血常规、尿常规、粪常规、血肌酐、血尿素氮、肝功能、血脂、血糖、糖化血红蛋白、电解质、肌酸激酶、脑钠肽、肌钙蛋白、D-二聚体、国际标准化比值、凝血酶原时间、凝血酶时间、部分凝血活酶时间、甲状腺功能及甲状腺抗体、同型半胱氨酸、叶酸、维生素B12、抗磷脂抗体(antiphospholipid antibody,APA)、抗溶血性链球菌“O”、血液免疫球蛋白组合(免疫球蛋白G、M、A)、类风湿因子、抗环瓜氨酸肽抗体、抗中性粒细胞胞浆抗体(MPO-ANCA、PR3-ANCA、p-ANCA、c-ANCA)、抗核抗体谱(抗 ds-DNA、抗SSA、抗SSB、抗U1RNP、抗Scl-70、抗Sm、抗核糖体P蛋白、抗Jo-1、抗Ro-52、抗着丝点蛋白B、抗核小体、抗组蛋白、抗线粒体M2型)、肿瘤标志物(CA199、CEA、AFP、CA125、CA211、CA724、NSE、SCC鳞 癌 抗 原、CA50、PSA、FPSA)、人类免疫缺陷病毒、梅毒血清学、乙肝表面抗原、丙肝抗体检测值均在正常范围内。腰椎穿刺检查:脑脊液压力为14 cm H2O(1 cm H2O=0.098 kPa),脑脊液常规、生化、乳胶凝集试验、免疫球蛋白、革兰染色、抗酸染色、墨汁染色结果均正常。血清及脑脊液中自身免疫性脑炎抗体、中枢系统脱髓鞘抗体、副肿瘤抗体均为(-)。
肺部高分辨率计算机断层成像(high resolution computed tomography,HRCT)显示双侧腋下多发淋巴结,脾脏饱满,余未见明显活动性病变。腹部B超检查显示脾肿大,余未见明显异常。颈部淋巴结超声检查显示双侧颈部可见数枚淋巴结,最大者位于颈上部(22 mm×8.6 mm),边界清,皮髓质分界清。头颅MRI平扫+增强扫描显示右侧侧脑室旁、延髓偏右侧小斑片状异常信号影,T2加权成像、液体衰减反转恢复(fluid attenuated inversion recovery,FLAIR)序列均呈稍高信号影,未见明显强化(图1B、C、D、E)。心电图、心脏彩超、双下肢动静脉超声、脑电图、肌电图、神经传导速度、脑干听觉诱发电位、视觉诱发电位、头颅磁共振血管成像以及颈椎、胸椎及眼MRI平扫+增强扫描均未见异常。
邀请本院耳鼻喉科会诊:右耳感音神经性耳聋,左耳正常。邀请本院眼科会诊:右眼视力为0.3,左眼视力为1.0,双眼向右视时复视,右眼外展露白2 mm,余结膜、角膜、前房、瞳孔、眼底视网膜和视盘未见明显异常。皮肤活检病理学检查结果(图3A、B、C):皮肤组织、真皮内及皮下中小动脉均未见血栓形成。
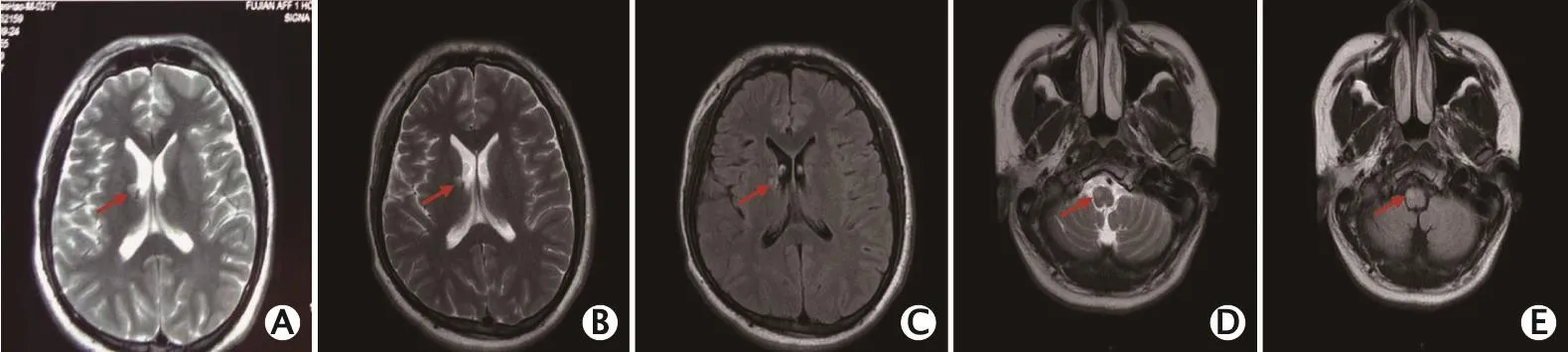
图1 1例诊断为Sneddon综合征(Sneddon’s syndrome,SS)的男性患者的头颅MRl。A(2015年9月22日):T2加权成像呈高信号影(红色箭头所示);B~E(2017年3月27日):右侧侧脑室旁、延髓偏右侧小斑片状异常信号影,T2加权成像、液体衰减反转恢复(fluid attenuated inversion recovery,FLAlR)序列均呈稍高信号影,未见明显强化(红色箭头所示)
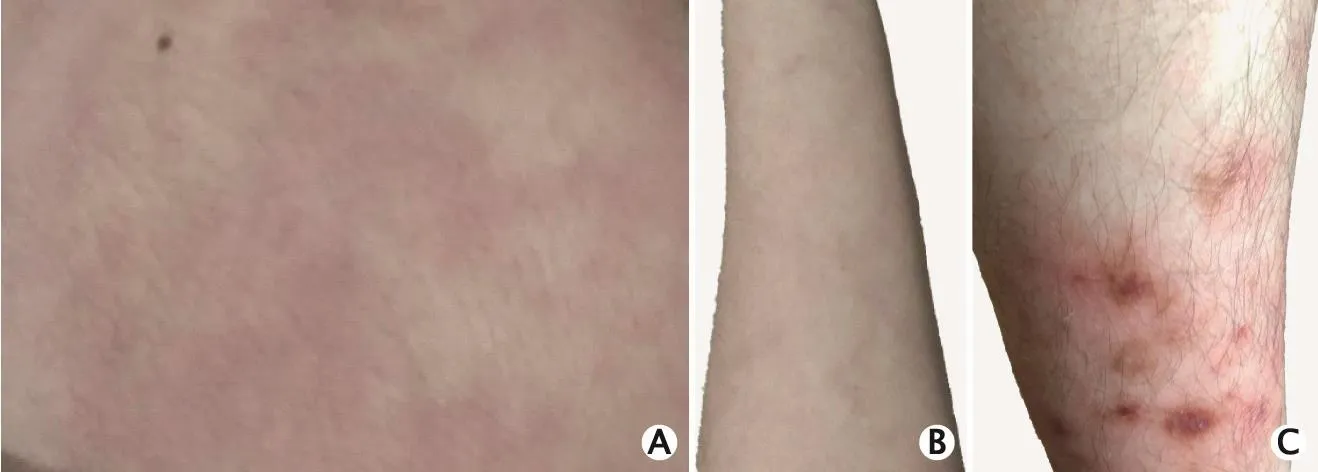
图2 1例诊断为Sneddon综合征(Sneddon’s syndrome,SS)的男性患者的皮肤表现。A、B、C依次为患者的手背、前臂和小腿,网状青斑表现为淡红色、蓝色、紫红色的不规则花边状及裂环状,部分破溃
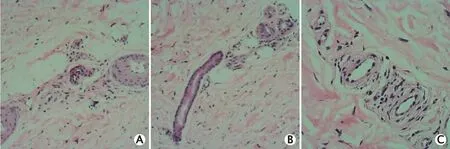
图3 1例诊断为Sneddon综合征(Sneddon’s syndrome,SS)的男性患者的皮肤活检病理学检查结果,可见皮肤组织、真皮内及皮下中小动脉无血栓形成。A:苏木精-伊红染色,×10;B:苏木精-伊红染色,×20;C:苏木精-伊红染色,×40
入院后,排除其他可导致皮肤网状青斑和(或)脑卒中发作的疾病,临床诊断为SS。给予患者阿司匹林抗血小板聚集,同时给予前列地尔改善循环、甲钴胺营养神经、碳酸氢钠片碱化尿液等对症支持治疗2周,患者皮肤网状青斑明显消退,头晕、复视、行走不稳症状亦有所好转。因患者尿酸水平偏高,遂改阿司匹林(100 mg/次,1次/d)为氯吡格雷(75 mg/次,1次/d)抗血小板治疗,并加用非布司他(40 mg/次,1次/d)降低尿酸水平。
出院1个月后随访,患者皮肤网状青斑无进一步加重,头晕、复视及行走不稳症状与出院时基本相似。随访9个月至今,患者皮肤网状青斑的严重程度偶因气温变化而稍有波动,头晕、复视症状基本消失,行走不稳症状基本同出院时。
2 讨 论
SS是一种罕见的全身系统性疾病,发病率约为4/1 000 000,80%为中青年女性患者[1]。SS的具体病因及发病机制尚未明确,病理改变为广泛累及中小动脉的非炎性、血栓性闭塞[2]。雌激素、口服避孕药、高血脂和高血压是SS发展的高危因素。APA阳性率为40%~50%[3-5],其水平与血液高凝状态密切相关,提示APA可能在SS的发展中起关键作用。SS主要为散发性,但也有少数家族病例的报道。ZHOU等[6]和NAVON等[7]的研究明确指出,腺苷脱氨酶2(adenosine deaminase 2,ADA2)基因突变可引发以皮疹、反复卒中和血管炎为主要表现的SS的发生。BRAS等[8]则报道,ADA2基因缺失可作为家族性SS的病因。
脑卒中发作是SS的主要临床表现之一,脑出血、蛛网膜下腔出血及脑室内出血在SS中较少见,90%以上为急性脑梗死或短暂性脑缺血发作;病灶大多位于大脑中动脉或大脑后动脉供血区域;最常见的神经系统症状为偏瘫、感觉障碍、失语、视力下降和视野缺失等,而头痛是最常见的非特异性主诉[9-11],亦有核间性眼肌麻痹、单侧动眼神经麻痹[12-13]。77%的SS患者有精神障碍,以抑郁、认知损害、注意力下降、记忆力减退、视觉感知及视空间构建异常最为常见[14-15]。皮肤网状青斑是SS的另一项主要临床表现,一般先于脑卒中发作前数年出现,也有少数病例晚于脑卒中发作出现,表现为淡红色、蓝色、紫红色的不规则花边状及裂环状,是皮肤血流障碍表现;多累及身体2个及以上部位,可分布于四肢(100%)、躯干(84%~89%)、臀部(68%~74%)、手足(53%~59%)、 面部(15%~16%);皮肤网状青斑一般持续存在,其颜色及范围可在寒冷、妊娠及神经功能急性损害期加重,严重时可出现溃疡[4,16]。皮肤活检的典型发现为皮下中小动脉进行性非炎性血栓形成,致血管管腔闭塞及代偿性毛细血管扩张,部分病例可见因内皮细胞及平滑肌细胞增生所致的闭塞性动脉内膜炎。皮肤活检具有诊断意义,但单次活检阳性率仅为27%[17]。除神经系统和皮肤表现以外,SS可伴有其他系统的损害,包括高血压(65%)、心脏瓣膜病(61%)、缺血性心脏病(50%~70%)、肾脏受累(50%~70%)、胃肠症状、贫血、静脉血栓形成及罕见的主动脉瘤样扩张等[18-21]。临床上,SS应与引起类似皮肤网状青斑改变和(或)反复缺血性卒中发作的其他疾病相鉴别,包括原发性APA综合征、Divry-VanBogert综合征、中枢神经系统血管炎、系统性红斑狼疮、结节性多动脉炎、冷球蛋白血症、青斑样血管病、冷凝集素病、胆固醇栓塞综合征、多发性硬化、多发性心源性脑栓塞、感染性疾病(如梅毒和莱姆病)等[1,3,5,22]。SS是一种临床综合征,其诊断主要基于皮肤网状青斑合并脑卒中发作,并排除其他可导致皮肤网状青斑和(或)脑卒中发作的疾病,典型的皮肤活检发现具有诊断意义[17,23]。SS的最佳治疗方案目前仍是未解之谜,基于目前对SS病因的认识,其治疗以抗血小板聚集或抗凝为主[24]。对于APA阳性SS患者,推荐长期使用华法林抗凝,并且控制INR≥3。对于APA阴性的SS患者,建议给予阿司匹林联合氯吡格雷抗血小板治疗,若为降低出血风险,则可考虑单用阿司匹林或氯吡格雷抗血小板治疗,也可尝试使用糖皮质激素和免疫抑制剂,但疗效并不确切。另有静脉使用前列地尔显著改善SS患者皮肤溃疡的报道[16]。目前,国内外关于SS预后的研究还较少。一项平均随访7.4年的研究报道,82%的SS患者未遗留明显的神经功能缺损[11];一项平均随访6.2年的研究报道,SS死亡率为9.5%[3]。
与大多数SS患者为中青年女性且先出现皮肤网状青斑略有不同,本病例为青年男性,以反复发生脑梗死为首发表现,头颅MRI(图2B、C、D、E)T2加权成像及FLAIR序列可见右侧侧脑室旁、延髓偏右侧小斑片状稍高信号影,符合反复脑梗死的临床诊断,且病灶部位与SS所致脑梗死病灶大多位于大脑中后动脉供血区域相吻合[11]。本病例的皮肤网状青斑广泛分布于躯干和四肢,表现为淡红色、蓝色、紫红色的不规则花边状及裂环状,部分破溃,虽迟于神经系统损害出现,但仍符合文献报道的SS皮肤损害的常见部位及形态学特点[4]。合并脾肿大及淋巴结肿大是本病例的特点,而SS合并脾肿大及淋巴结肿大的病例报道,既往仅有1例。2016年,BUKAVINA等[25]报道了1例37岁的女性,以广泛的皮肤网状青斑、慢性偏头痛、脾肿大及淋巴结肿大为主要临床表现。本病例虽因个人原因而未行淋巴结活检,但完善其他检查后初步排除可导致脾肿大及淋巴结肿大的常见血液系统疾病、肿瘤性疾病及感染性疾病,其脾肿大及淋巴结肿大仍考虑为SS所致。由此提示,网状内皮系统累及可能也是SS的特征之一,但具体机制不明,可能与某种攻击网状内皮系统的抗体有关,这种抗体不同于APA,且现有的实验室检查尚无法进行检测,应予以重视。本病例有右耳耳鸣和耳聋病史,脑干听觉诱发电位正常,耳鼻喉科会诊意见为右耳感音神经性聋。迄今为止,尚未见SS合并听力受损的病例报道。因蜗神经核团由双侧大脑半球支配,因此单侧脑梗死并不会导致听力下降。本病例出现右侧感音神经性聋,可能与供应右侧蜗神经核团以下听觉通路的微小血管受累有关。本病例的MMSE量表评分为26分,结合其大专文化水平,评估为存在认知功能轻度下降,随着病情的进展,有进展为血管性痴呆的可能。本病例于入院前1周有头痛史,亦与SS常见的非特异性主诉相符。本病例还否认既往有高血压、肾脏病史,但入院后多次检查发现血压升高、尿酸水平升高,推测可能与疾病所致血管及肾脏损害相关。本病例的C反应蛋白水平和红细胞沉降率升高,但缺少导致这2项指标异常的其他病因依据,因此推测可能与SS处于急性期有关。本病例皮肤活检未发现典型的病理学改变。文献报道单次皮肤活检的阳性率不高;而随着活检次数的增加,其阳性率可逐步提高,2次活检的阳性率可达53%,3次活检的阳性率达80%[17]。本病例拒绝再次进行皮肤活检。在排除其他可能导致皮肤网状青斑和(或)脑卒中发作的疾病后,本病例被临床确诊为SS;给予抗血小板和改善循环治疗的疗效确切。
SS作为一种罕见的累及多系统的神经皮肤综合征,当症状不典型或表现为非特异症状时,易发生误诊和漏诊。随着脑卒中的反复发生,SS发生严重神经功能缺损及血管性痴呆的可能性增加。因此,早期识别SS至关重要,以便尽早地做出正确诊断并给予治疗,从而改善SS患者的预后。
[1]WU S,XU Z,LlANG H.Sneddon's syndrome:a comprehensive review of the literature[J/OL].Orphanet J Rare Dis,2014,9:215(2014-12-31)[2018-02-01].DOl:10.1186/s13023-014-0215-4.
[2]HlLTON DA,FOOTlTT D.Neuropathological findings in Sneddon's syndrome[J].Neurology,2003,60(7):1181-1182.
[3]ZELGER B,SEPP N,STOCKHAMMER G,et al.Sneddon’s syndrome.A long-term followup of 21 patients[J].Arch Dermatol,1993,129(4):437-447.
[4]FRANCÈS C,PAPO T,WECHSLER B,et al.Sneddon syndrome with or without antiphospholipid antibodies.A comparative study in 46 patients[J].Medicine(Baltimore),1999,78(4):209-219.
[5]FETONl V,GRlSOLl M,SALMAGGl A,et al.Clinical and neuroradiological aspects of Sneddon’s syndrome and primary antiphospholipid antibody syndrome.A follow-up study[J].Neurol Sci,2000,21(3):157-164.
[6]ZHOU Q,YANG D,OMBRELLO AK,et al.Earlyonset stroke and vasculopathy associated with mutations inADA2[J].N Engl J Med,2014,370(10):911-920.
[7]NAVON ELKAN P,PlERCE SB,SEGEL R,et al.Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy[J].N Engl J Med,2014,370(10):921-931.
[8]BRAS J,GUERRElRO R,SANTO GC.MutantADA2 in vasculopathies[J].N Engl J Med,2014,371(5):478-480.
[9]BOESCH SM,PLÖRER AL,AUER AJ,et al.The natural course of Sneddon syndrome:clinical and magnetic resonance imaging findings in a prospective six year observation study[J].J Neurol Neurosurg Psychiatry,2003,74(4):542-544.
[10]BAClU P,NOFAR CM,SPAULDlNG J,et al.Branch retinal artery occlusion associated with paracentral acute middle maculopathy in a patient with livedo reticularis[J].Retin Cases Brief Rep,2017,11(4):356-360.
[11]BOTTlN L,FRANCÈS C,DE ZUTTERE D,et al.Strokes in Sneddon syndrome without antiphospholipid antibodies[J].Ann Neurol,2015,77(5):817-829.
[12]JlMÉNEZ-GALLO D,ALBARRÁN-PLANELLES C,LlNARES-BARRlOS M,et al.Sneddon syndrome presenting with unilateral third cranial nerve palsy[J].J Neuroophthalmol,2014,34(1):50-52.
[13]REHANY U,KASSlF Y,RUMELT S.Sneddon’s syndrome: neuro-ophthalmologic manifestations in a possible autosomal recessive pattern[J].Neurology,1998,51(4):1185-1187.
[14]WElSSENBORN K,RÜCKERT N,EHRENHElM C,et al.Neuropsychological deficits in patients with Sneddon’s syndrome[J].J Neurol,1996,243(4):357-363.
[15]HSU FF,CHUNG KH.Psychosis with suicide attempt in Sneddon syndrome[J].Psychiatry Clin Neurosci,2017,71(2):147-148.
[16]COLLANTES-RODRÍGUEZ C,JlMÉNEZ-GALLO D,ARJONA-AGUlLERA C,et al.Treatment of skin ulcers secondary to Sneddon syndrome with alprostadil (prostaglandin e1)[J].JAMA Dermatol,2016,152(6):726-727.
[17]WOHLRAB J,FlSCHER M,WOLTER M,et al.Diagnostic impact and sensitivity of skin biopsies in Sneddon’s syndrome.A report of 15 cases[J].Br J Dermatol,2001,145(2):285-288.
[18]FRANCÈS C,PlETTE JC.The mystery of Sneddon syndrome: relationship with antiphospholipid syndrome and systemic lupus erythematosus[J].J Autoimmun,2000,15(2):139-143.
[19]KlRSANOVA TV,KOZLOVSKAlA NL,KALASHNlKOVA LA,et al.Characteristics of the kidney lesion in a patient with Sneddon's syndrome[J].Ter Arkh,2009,81(8):73-77.
[20]KRAEMER M,BAUMGAERTEL MW,BERLlT P.Miscarriage,peripheral thromboses and aortic aneurysm in antiphospholipid-antibodynegative Sneddon's syndrome[J].J Neurol,2007,254(11):1599-1600.
[21]DOMlNGUEZ F,PlESKE B,KELLE S.Cardiac manifestations of Sneddon's syndrome[J].lnt J Cardiol,2015,190:275-276.
[22]BERSANO A,MORBlN M,ClCERl E,et al.The diagnostic challenge of Divry van Bogaert and Sneddon Syndrome: Report of three cases and literature review[J].J Neurol Sci,2016,364:77-83.
[23]ClRlLLO G,TESSlTORE A,ClRlLLO M,et al.Livedo and ischemic strokes: diagnostic hints of a rare condition[J].Neurol Sci,2013,34(11):2073-2075.
[24]FLÖEL A,lMAl T,LOHMANN H,et al.Therapy of Sneddon syndrome[J].Eur Neurol,2002,48(3):126-132.
[25]BUKAVlNA L,WEAVER J,NAGY T,et al.ldiopathic livedo racemosa presenting with splenomegaly and diffuse lymphadenopathy[J].Cutis,2016,98(4):E26-E29.
猜你喜欢
世界科学技术-中医药现代化(2022年2期)2022-05-25
昆钢科技(2022年1期)2022-04-19
天津医科大学学报(2021年4期)2021-08-21
世界科学技术-中医药现代化(2021年12期)2021-04-19
作文评点报·低幼版(2020年25期)2020-07-23
养生保健指南(2019年11期)2019-12-17
中国感染与化疗杂志(2018年3期)2018-01-20
郑州大学学报(医学版)(2015年1期)2015-02-27
右江医学(2014年1期)2014-03-22
右江医学(2014年1期)2014-03-22
