
Development and validation of InDel markers for identification of QTL underlying flowering time in soybean
2018-04-12 03:33JilinWngLingpingKongKnchoYuFenggeZhngXinyiShiYnpingWngHiyngNnXiohuiZhoSijiLuDongCoXiomingLiChoFngFeifeiWngTongSuShichenLiXiohuiYunBohuiLiuFnjingKong
The Crop Journal 2018年2期
Jilin Wng,Lingping Kong,Kncho Yu,Fengge Zhng,Xinyi Shi,Ynping Wng,Hiyng Nn,Xiohui Zho,b,Siji Lu,b,Dong Co,Xioming Li,e,Cho Fng,e,Feifei Wng,e,Tong Su,e,Shichen Li,e,Xiohui Yun,*,Bohui Liu,b,**,Fnjing Kong,b,**
a The Key Laboratory of Soybean Molecular Design Breeding,Northeast Institute of Geography and Agroecology,Chinese Academy of Sciences,Harbin 150081,Heilongjiang,China
b School of Life Sciences,Guangzhou University,Guangzhou 510006,Guangdong,China
c Qiqihar Branch of Heilongjiang Academy of Agricultural Sciences,Qiqihar 161006,Heilongjiang,China
d Mudanjiang Branch of Heilongjiang Academy of Agricultural Sciences,Mudanjiang 157041,Heilongjiang,China
e University of Chinese Academy of Sciences,Beijing 100049,China
f College of Agriculture,Northeast Agricultural University,Harbin 150030,Heilongjiang,China
1.Introduction
A functional gene can be identified via forward and reverse genetics strategies[7,8].Positional cloning is widely used as a forward genetics approach to isolate genes in different organisms[9],and its utility can be fully exploited in modern molecular plant breeding systems,such as corn and soybean,when markers linked to genes of interest are discovered[10].The principle of positional cloning is to systematically narrow down the genetic interval containing a causal mutation by sequentially excluding all other regions in the genome[11].All rely on the development of highly dense genetic markers that are polymorphic between the accessions used for generating the mapping population(s)to provide adequate mapping resolution.This dependence is a major limiting factor for the rate of mapping progress.
With the decreasing cost of next-generation sequencing,there have been several proposals to exploit single-nucleotide polymorphisms(SNPs)and Insertion/Deletions(InDels)for genetic mapping with high-density markers.In contrast to SNPs,InDel polymorphisms,another form of natural genetic variation,have received relatively little attention.Mechanisms such as transposable elements,slippage in simple sequence replication,and unequal crossover events can result in the formation of InDels[12].They can be converted to a user-friendly marker type,show high variation and codominant inheritance,and are relatively abundant and uniformly distributed throughout the genome[13,14].InDel markers are PCR-based and readily genotyped by fragment length polymorphism with minimal laboratory equipment.Recently InDel markers have been widely applied for genotyping,genetic diversity analysis,QTL mapping,map-based cloning,and even marker-assisted selection in Arabidopsis,rice,wheat,turnip,sunflower,pepper,sesame,cotton,and citrus[14–27].However,InDel markers have seldom been identified and used in soybean.A recent study used 73,327 InDels in six soybean cultivars to build a soybean barcode system for comparing data from different sources[28].In another study,165 validated InDel markers were used to develop an InDel-based linkage map for a mapping population between Hedou 12 and Williams 82[29].By exploiting the reference genome sequence of soybean and the large amount of intensive resequencing data available in public databases[30–35],it is now possible to detect genome-wide InDel polymorphisms amongst different accessions using whole-genome resequencing to guide rapid and efficient development of InDel markers for high-resolution genetic analysis.
In this study,we attempted to develop InDel markers using genomic resequencing data using a series of bioinformatic approaches.In total,these methods yielded 12,619 new markers that were variously polymorphic amongst 56 soybean accessions.An InDel-based genetic map of soybean was constructed with 300 polymorphic InDel markers.QTL analysis was performed to identify genomic regions associated with flowering time.One major QTL(qDTF4)was identified in 2015 and confirmed in 2016.The InDel markers,genetic map,and QTL identified in this study will lay a foundation for the genetic/QTL analysis and isolation of genes underlying variation in flowering time and provide useful information for MAS breeding in soybean.
2.Materials and methods
2.1.Plant materials and trait evaluation
The F7:8seeds for the mapping populations were grown in walk-in plant growth chambers at 22°C,65%relative humidity,and long-day(LD)photoperiod(16 h light/8 h dark)in October 2015 and in the field in Harbin(45°43′N,126°45′E)and Mudanjiang(44°36′N,129°35′E),China in May 2016.
Days to flowering were recorded at the R1 stage(days from emergence to first open flower appearing on 50%of plants).For chamber experiments,seeds from each line were sown in pots.After germination,the seedlings were thinned until each pot contained five uniform plants.Populations were sown in the field with a single seed every 20 cM in 5-m rows spaced 60 cM apart and 25 seeds per line.All trials received standard cultural practices to control insects and weeds.
2.2.Mapping populations and sequence data sets
The BA population,derived from a cross between Mufu12-604×HB-2 and consisting of 156 F2genotypes,was used to test the newly developed markers and construct a high-density InDel linkage map.The DW population(144 RILs),derived from a cross between Dongnong 50(early-flowering in LD photoperiod)and Williams 82(late-flowering in LD photoperiod),was used to evaluate the InDel markers for QTL mapping.
Fifty six accessions,including 29 from three recent research papers and 27 from this study,were used for InDel polymorphism validation(Table 1).Young leaves from 27 accessions were collected three weeks after planting in growth chambers and separately quick-frozen in liquid nitrogen.Total DNA was extracted by the improved cetyltrimethylammonium bromide(CTAB)method[36].A sequencing library was constructed with at least 6 μg of genomic DNA following the manufacturer's instructions(Illumina Inc.,San Diego,CA).Paired-end sequencing libraries with an insert size of approximately 500 bp were sequenced on an Illumina HiSeq 2000 sequencer.
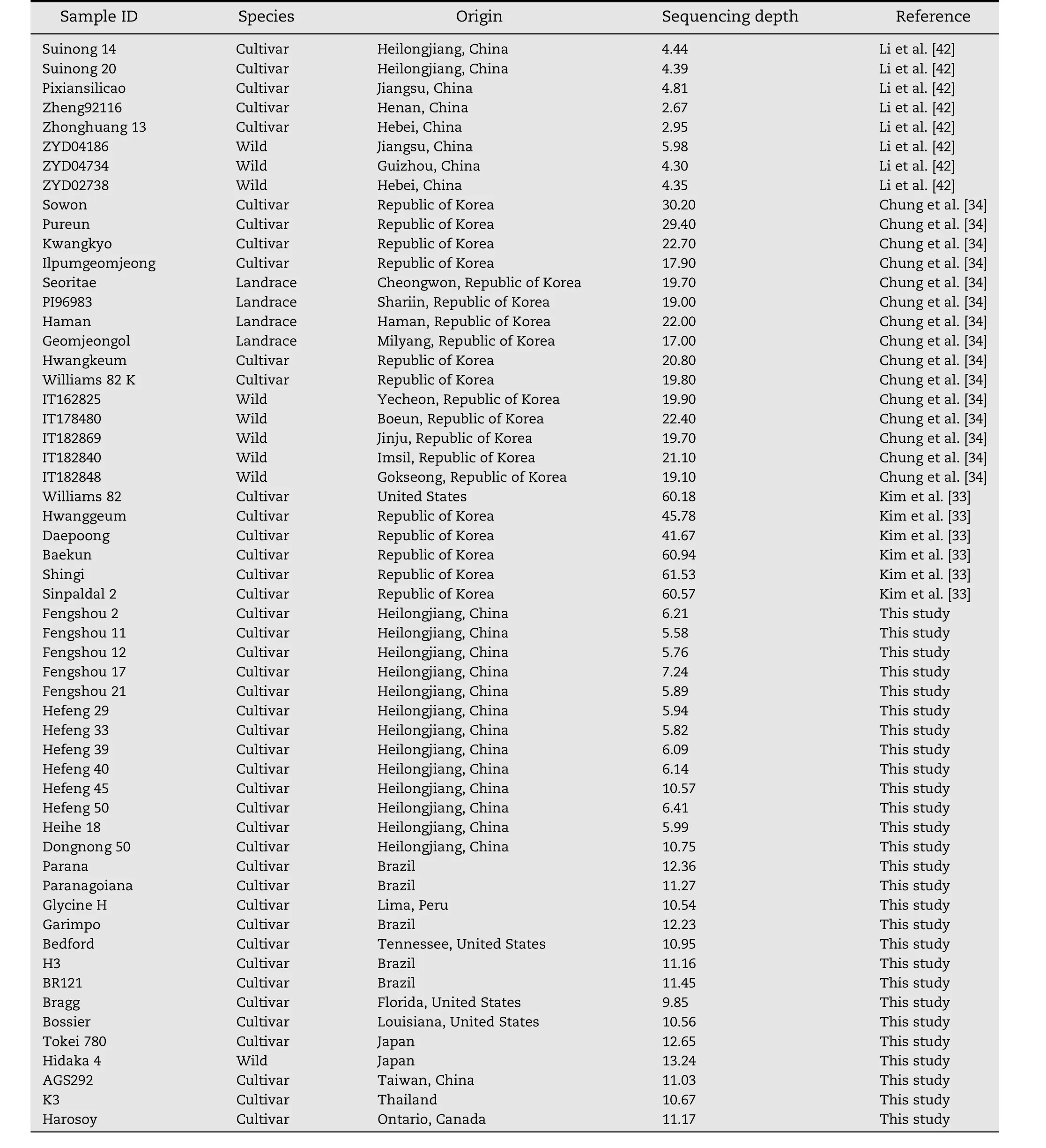
Table 1–Soybean accessions used in the study.
2.3.InDel detection and marker development
The process used to detect InDel sites involved three steps.(i)Alignment of paired-end (PE) short reads. BWA(Burrows-Wheeler Aligner)software[37]was used to align paired reads to the reference genome with default parameters and Picard(http://broadinstitute.github.io/picard/)to mark duplicate reads.(ii)Detection of InDels.Five software tools:Samtools[38],GATK Unique Genotyper[39],Varscan[40],Pindel[41],and Soapindel[42],were used to identify InDels 5–50 bp in length.(iii)Optimization of InDels.A support vector machine(SVM)filter was trained on simulated data using a library for support vector machines(LIBSVM)[43]and the InDels were filtered with the SVM filter.The InDels with high polymorphism(MAF>0.4)among 56 individuals were chosen as molecular markers.
此时养生应遵循阳气潜藏的规律,以“养藏”为根本,适当进补,所谓“立冬补一冬”。冬令进补,要注意一个“藏”字,达到敛阴护阳、养精蓄锐的目的。冬补以炖补为佳,炖补制作时间长,有利于营养消化吸
Primer 3 software[44]was employed to identify primers for each InDel site with the following parameters:predicted products ranged from 100 to 300 bp;the length of primers was limited to 18–24 bp with an optimum size of 20 bp;the annealing temperature was restricted to 57–62 °C;the GC content was set to 35%,50%,and 65%as the minimum,optimum,and maximum,respectively.Only primers with one hit in the genome assembly were retained.
2.4.Nomenclature
In order to provide the user with valuable information on marker distribution,the markers were named using the format IDNNXXXX,where ID represents InDel,NN the chromosome number(01–20),and the Xs the ordered number of each marker on its chromosome.For example,InDel marker ID06006 is the sixth marker on chromosome Gm06.
2.5.Screening and genotyping of InDel markers
Total genomic DNA was extracted from young leaves or seed flour of individual samples using the improved CTAB method.PCR amplification was performed in a 10 μL reaction consisting of a final concentration of 1×Easy Taq PCR SuperMix for PAGE(TransBionovo Co.,Ltd.,Beijing,China),0.2 μmol L−1forward/reverse primers,and approximately 30–50 ng of genomic DNA as a template.The amplification protocol comprised an initial denaturation for 2 min at 94°C,35 cycles of denaturation for 30 s at 94°C,annealing for 30 s at 56 °C,and extension for 30 s at 72 °C,followed by a final extension for 5 min at 72°C.PCR products were resolved by 12%SDS-polyacrylamide gel electrophoresis.The gels were stained with ethidium bromide,and the bands were visualized and photographed under ultraviolet light.
2.6.Construction of a linkage map and QTL analysis of flowering time
The F2population,BA,was used to evaluate the utility of InDel makers for mapping.JoinMap 4.0[45]was used to build the genetic map with 347 markers that were polymorphic between the two parents.The groups and orders of segregated markers were determined on the basis of an LOD(logarithm of the odds ratio for linkage)score of≥7.0 and a minimum LOD score of 1.0,with the threshold of 0.4 in each LG.Markers were tested for deviation from expected Mendelian segregation using a chi-squared test and sorted on the basis of the test(P<0.05).Both inclusive composite interval mapping(ICIM)and multiple-QTL mapping(MQM)were initially applied to detect QTL(LOD>2.0)for flowering time,using QTL IciMapping 4.0[46]and MapQTL 5[47],respectively.
3.Results
3.1.InDel identification and marker development in 56 soybean accessions
Many accurate strategies with corresponding cost and throughput have been developed to detect SNPs as new polymorphic markers for the success of a map-based cloning project.However,detecting InDels is a more challenging task and requires substantial bioinformatic analysis.Several factors affect the discovery of InDels.The phylogenetic relationship between the genotypes used for InDel discovery is important.In this study,based on the alignment of the sequencing reads to a reference genome,17,613 InDel sites were identified among 56 soybean accessions including nine wild soybeans,four landraces,and 43 cultivars from many countries(Table 1).
The InDel sites were filtered by size and those with a size of 5–50 bp were retained.In total,12,619 primer pairs were obtained with a dense distribution across each of the 20 soybean chromosomes(Table S1).The frequency of InDel markers varied across the chromosomes,falling within the range of approximately 275–1207 markers per chromosome(Table 2).Based on this distribution of InDel markers,it was possible to construct high-density genetic maps and select InDels within specific regions for fine mapping.
To evaluate the performance of the InDel markers,1000 random markers were tested by PCR with Williams 82 as the template.A total of 930 markers(93%)generated single and clear bands as expected,and only 70 markers(7%)either yielded no amplification product or were difficult to score.We next examined the distribution of the 12,619 InDels relative to genes of soybean and found that 429(3.4%)were located within the exons of annotated genes,where gene function may be expected to be influenced.Of these,135(1.1%)were non-3-nucleotide InDels,which were predicted to cause frameshift mutations.This finding indicates that the developed InDel markers are useful for identifying the genetic composition of soybean and provide a valuable source of allelic diversity for genetic and molecular dissection of traits.
3.2.Genetic map construction
The developed InDel markers should be useful for genetic map construction because there are on average about 630 markers on each chromosome.We used a F2mapping population to illustrate their application to linkage analysis.The F2population consisted of 156 progeny derived from the cross Mufu 12-604×HB-2,which were not included in the 56 soybean accessions.A random subset of 2841 primer pairs were chosen to identify polymorphism between the parental lines,and 347(12.21%)polymorphic markers were validated.This finding shows that these InDel markers have universal applicability of performance and application,and can be expanded to all soybean germplasm,although these InDel markers were designed to capture the variation within 56 soybean accessions.
A total of 347 polymorphic markers were scored in the genotype analysis of 156 progeny in the BA F2population,with each primer pair yielding polymorphic bands at a single locus.After exclusion of 47 unlinked markers,300 marker loci were grouped into 20 LGs,which matched the 20 consensus LGs.Finally,a genetic map(Fig.1),designated as the BA map,was constructed with 20 LGs covering a total genetic distance of 2347.30 cM with an average density of one marker for every 7.82 cM(Table 2).The number of mapped markers per LG ranged from 10(H and D2)to 23(A2)with an average of 15 markers.The largest and smallest genetic distances between adjacent markers were 52.3 cM and 0.1 cM,respectively.Because of low marker density(Fig.2)and infrequent recombination compared with distal regions,our map did not cover all centromeric blocks,resulting in coverage of only a portion of some chromosomes(N,C2,M,O,H,and F)or of two clusters of markers,one from each arm(K and B1)in the F2mapping study.Six marker orders(N,A1,M,B2,and E)in our genetic map that were in conflict with the physical map could be due to sequence assembly errors,inversions,and segregation distortion.
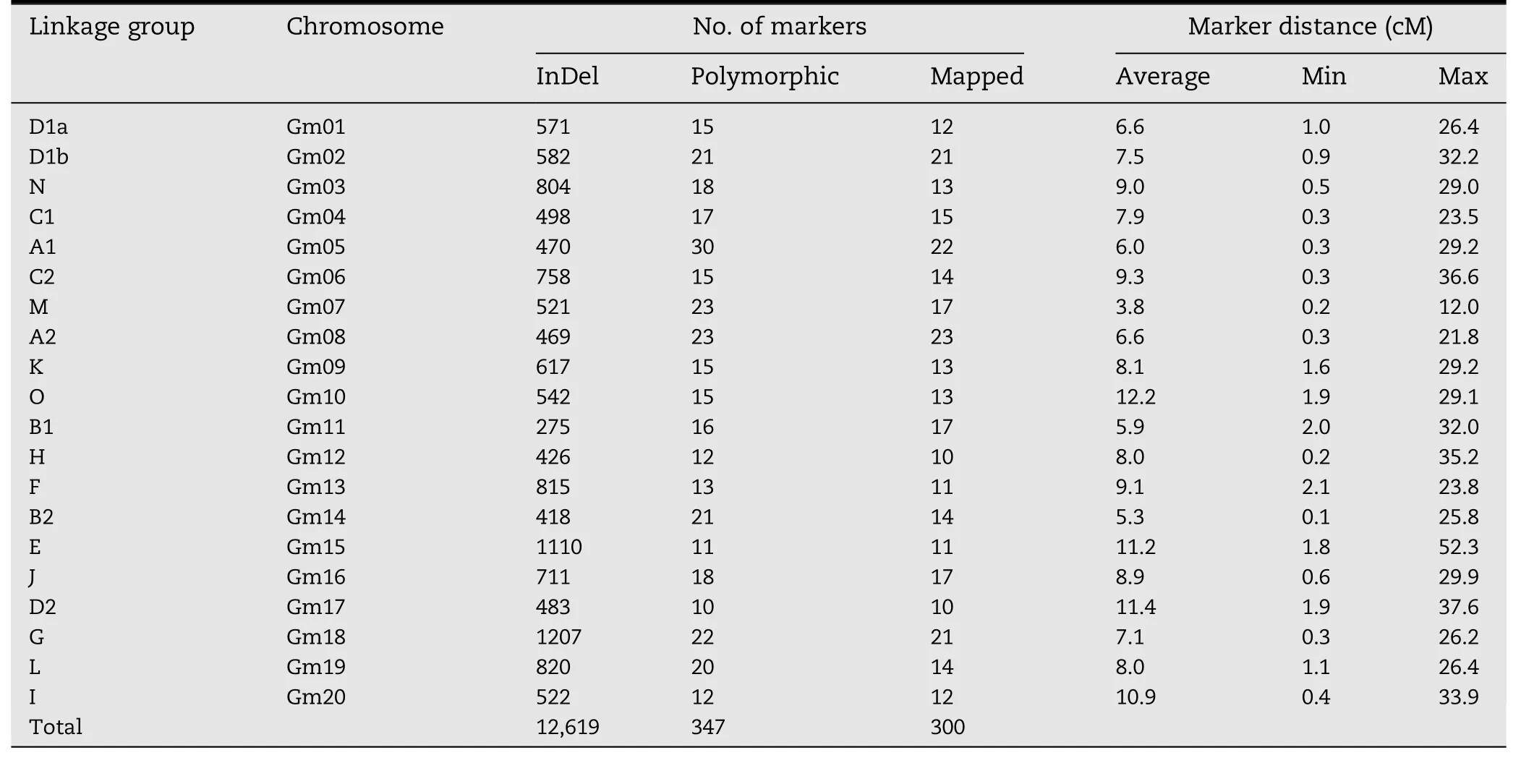
Table 2–Statistics of the BA map based on InDel markers.
3.3.QTL analysis of flowering time
The DW population(144 RILs)originated from a cross between the Chinese cultivar Dongnong 50 and the American cultivar Williams 82 and was used to evaluate the InDelmarkers for QTL mapping.The F7:8seeds were grown in walk-in plant growth chambers in October 2015.A total of 4 QTL,including one major(qDTF4)and three minor QTL(qDTF20,qDTF13,and qDTF12),were detected on four chromosomes using either ICIM or MQM.These QTL explained from 6.0%to 11.3%of phenotypic variation(PEV),with LOD scores ranging from 2.09 to 2.93(Table 3).
To confirm the QTL results,the F7:9seeds were grown in the field in Harbin and Mudanjiang on May 2016.The major QTL,which was assumed to be identical to qDTF4,was repeatedly identified by both ICIM and MQM in two environments.This result showed that the effect of qDTF4 was little affected by the environment and was consistent with the characterization of high heritability of flowering time.In addition,another minor QTL(qDTF11)was mapped on chromosome 11,and explained 6.5%and 9.4%of the phenotypic variation,with LOD scores 2.58 and 3.31,by ICIM and MQM,respectively(Table 3).
4.Discussion
Genetic diversity in soybean as in other crops has decreased during domestication and improvement[35].The phylogenetic relationship between the genotypes used for InDel discovery is important.In this study,we collected 56 soybean accessions from several regions around the world,including nine wild soybeans,four landraces,and 43 cultivars.The germplasm from wild soybeans and landraces would therefore be useful in broadening the genetic basis and the detection of InDels.This report presents an optimized algorithm with no special requirements for the number of accessions and InDel detection software tools.Additional software can be added to this InDel detection procedure to further improve the performance of the proposed algorithms.

Fig.1–Genetic linkage map of soybean constructed with InDel markers.Genetic positions and marker names are indicated on the left and right side of each chromosome,respectively.
InDels identification has become routine with the abundance of next-generation sequence data.The InDel markers developed in this study could be widely used in genotyping with minimum lab equipment and PCR options.The potential utility of InDel markers in multiplex PCR could reduce the cost of genotyping by reducing the quantity of reagents and DNA in PCR reactions.Furthermore,this strategy is efficient when hundreds of markers are screened but DNA availability is limited.Our InDel markers closely match many of the criteria for multiplex PCR.The critical parameters of the primers in multiplex PCR should be 18–34 bp or more in length,GC content of 35%–60%,and annealing at 55–58 °C.In addition,the primer length should be up to 28–30 bp and the annealing temperature should be increased for reducing non-specific PCR products.However,owing to the finite polymerase and DNA resources,many specific loci strongly suppress non-specific amplification.Thus,54°C is the appropriate temperature for amplifying multiple loci at the same time[48].All primers reported here were designed with a length of 18–24 bp and GC content of 35%–65%and were amplified at 56 °C,indicating the potential utility of these markers for multiplex PCR.
Mapping QTL requires a genetic map covered with a high density of polymorphic markers.However,although reduction in the cost of next-generation sequencing technologies will allow the sequencing of numerous soybean accessions,the specialized expertise and the skilled applications of bioinformatics analysis will become a rate-limiting step in uncovering the molecular basis of natural variation.To avoid map-based cloning,a tedious task beset with complications,several recent papers have reported workflows for next-generation sequencing-based strategies for mutation mapping.The approach we advocate here is using resequenced genomes to rapidly facilitate InDel marker design for application to conventional mapping.Interestingly,Dongnong 50 and Williams 82 carry the same genotype(e1-as/E3/E4)for known major maturity loci,but a large difference of 30 days in R1 between the two cultivars was observed under long-day conditions.Thus,some new genes may be involved in control of flowering time and be strongly associated with photoperiod response.The main-effect QTL(qDTF4)was located in the same region as the E8 locus[49,50]and contained candidate genes E1-like-a and E1-like-b,two E1 homologs,which function similarly to E1 in adjusting flowering time in soybean[51].The frequencies of InDel markers developed in this study varied over chromosomes,falling within the range of 275–1207 markers per chromosome,indicating that it was possible to construct high-density genetic maps and select InDels within specific regions for fine mapping.
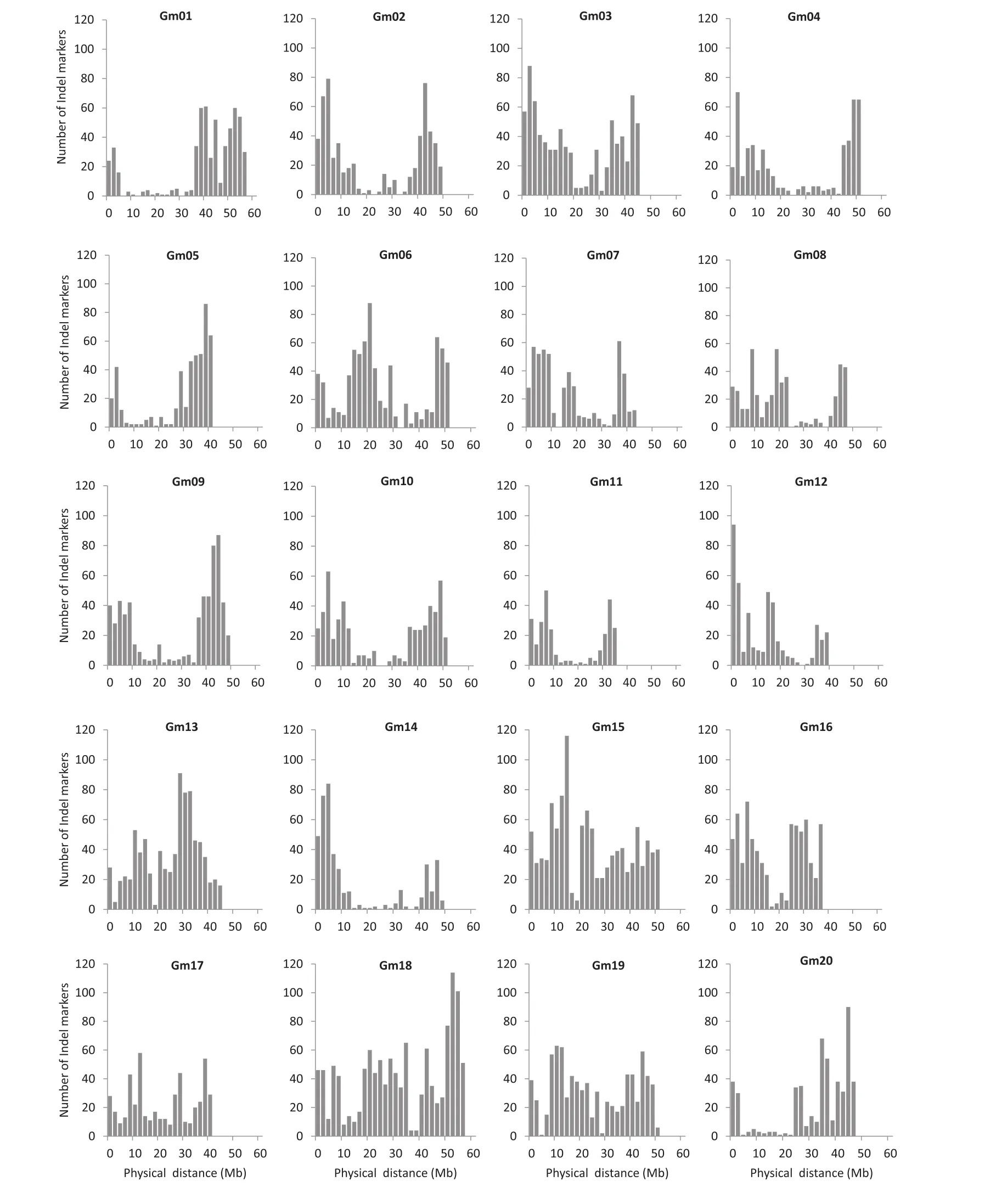
Fig.2–Physical distribution of 12,619 InDel markers across 20 chromosomes of soybean.The x axis shows the chromosome length in Mbp and the y axis the frequency of InDel markers.
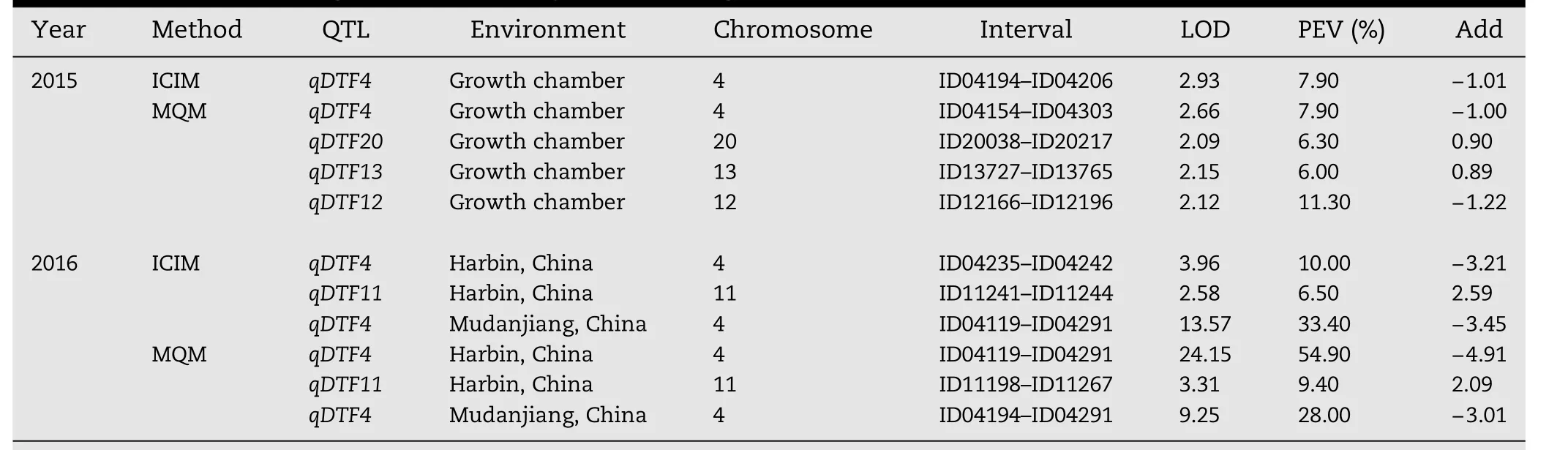
Table 3–QTL of flowering time identified by two mapping methods.
Supplementary data for this article can be found online at https://doi.org/10.1016/j.cj.2017.08.001.
This work was supported by National Natural Science Foundation of China(31430065,31571686,31371643,31071445),National Key Research and Development Program(2016YFD0100401),“Strategic Priority Research Program”of the Chinese Academy of Sciences(XDA08030108),the Open Foundation of the Key Laboratory of Soybean Molecular Design Breeding of Chinese Academy of Sciences,“One-hundred Talents”Startup Funds from Chinese Academy of Sciences,Scientific Research Foundation for Returned Chinese Scholars of Heilongjiang Province,China(LC201417),and the Science Foundation for Creative Research Talents of Harbin Science and Technology Bureau,China(2014RFQYJ046).
[1]R.G.Palmer,T.C.Kilen,Qualitative genetics and cytogenetics,in:J.R.Wilcox(Ed.),Soybeans:Improvement,Production,and Uses,Agronomy Monographs,2nd Edition,No.16,American Society of Agronomy,Crop Science Society of America,Soil Science Society of America, Madison, Wisconsin, USA 1987,pp.135–209.
[2]P.Keim,B.W.Diers,T.C.Olson,R.C.Shoemaker,RELP mapping in soybean:association between marker loci and variation in quantitative traits,Genetics 126(1990)735–742.
[3]J.G.K.Williams,A.R.Kubelik,K.J.Livak,J.A.Rafalski,S.V.Tingey,DNA polymorphisms amplified by arbitrary primers are useful as genetic markers,Nucleic Acids Res.18(1990)6531–6535.
[4]M.S.Akkaya,A.A.Bhagwat,P.B.Cregan,Length polymorphisms of simple sequence repeat DNA in soybean,Genetics 132(1992)1131–1139.
[5]M.S.Akkaya,R.C.Shoemaker,J.E.Specht,A.A.Bhagwat,P.B.Cregan,Integration of simple sequence repeat DNA markers into a soybean linkage map,Crop Sci.35(1995)1439–1445.
[6]Q.J.Song,G.F.Jia,Y.L.Zhu,D.Grant,R.T.Nelson,E.Y.Hwang,D.L.Hyten,P.B.Cregan,Abundance of SSR motifs and development of candidate polymorphic SSR markers(BARCSOYSSR_1.0)in soybean,Crop Sci.50(2010)1950–1960.
[7]J.M.Alonso,J.R.Ecker,Moving forward in reverse:genetic technologies to enable genome-wide phenomic screens in Arabidopsis,Nat.Rev.Genet.7(2006)524–536.
[8]C.Alonso-Blanco,M.G.Aarts,L.Bentsink,J.J.Keurentjes,M.Reymond,D.Vreugdenhil,M.Koornneef,What has natural variation taught us about plant development,physiology,and adaptation?Plant Cell 21(2009)1877–1896.
[9]X.F.Chi,X.Y.Lou,Q.Y.Shu,Progressive fine mapping in experimental populations:an improved strategy toward positional cloning,J.Theor.Biol.253(2008)817–823.
[10]H.A,Yang,Y.Tao,Z.Q.Zheng,C.D.Li,M.W.Sweetingham,J.G.Howieson,Application of next-generation sequencing for rapid marker development in molecular plant breeding:a case study on anthracnose disease resistance in Lupinus angustifolius L,BMC Genomics 13(2012)318.
[11]W.Lukowitz,C.S.Gillmor,W.R.Scheible,Positional cloning in Arabidopsis.Why it feels good to have a genome initiative working for you,Plant Physiol.123(2000)795–805.
[12]R.J.Britten,L.Rowen,J.Williams,R.A.Cameron,Majority of divergence between closely related DNA samples is due to indels,Proc.Natl.Acad.Sci.U.S.A.100(2003)4661–4665.
[13]R.E.Mills,C.T.Luttig,C.E.Larkins,A.Beauchamp,C.Tsui,W.S.Pittard,S.E.Devine,An initial map of insertion and deletion(INDEL)variation in the human genome,Genome Res.16(2006)1182–1190.
[14]D.I.Pacurar,M.L.Pacurar,N.Street,J.D.Bussell,T.I.Pop,L.Gutierrez,C.Bellini,A collection of INDEL markers for mapbased cloning in seven Arabidopsis accessions,J.Exp.Bot.63(2012)2491–2501.
[15]X.Hou,L.Li,Z.Peng,B.Wei,S.Tang,M.Ding,J.Liu,F.Zhang,Y.Zhao,H.Gu,L.J.Qu,A platform of high-density INDEL/CAPS markers for map-based cloning in Arabidopsis,Plant J.63(2010)880–888.
[16]K.Hayashi,H.Yoshida,I.Ashikawa,Development of PCR-based allele-specific and InDel marker sets for nine rice blast resistance genes,Theor.Appl.Genet.113(2006)251–260.
[17]P.Liu,X.X.Cai,B.R.Lu,Single-seeded InDel fingerprints in rice:an effective tool for indica-japonica rice classification and evolutionary studies,J.Syst.Evol.50(2012)1–11.
[18]H.Raman,R.Raman,R.Wood,P.Martin,Repetitive indel markers within the ALMT1 gene conditioning aluminium tolerance in wheat(Triticum aestivum L.),Mol.Breed.18(2006)171–183.
[19]B.Liu,Y.Wang,W.Zhai,J.Deng,H.Wang,Y.Cui,F.Cheng,X.W.Wang,J.Wu,Development of InDel markers for Brassica rapa based on whole-genome re-sequencing,Theor.Appl.Genet.126(2013)231–239.
[20]H.H.Lv,L.M.Yang,J.G.Kang,Q.B.Wang,X.W.Wang,Z.Y.Fang,Y.M.Liu,M.Zhuang,Y.Y.Zhang,Y.Lin,Y.H.Yang,B.Y.Xie,B.Liu,J.S.Liu,Development of InDel markers linked to Fusarium wilt resistance in cabbage,Mol.Breed.32(2013)961–967.
[21]A.Heesacker,V.K.Kishore,W.X.Gao,S.X.Tang,J.M.Kolkman,A.Gingle,M.Matvienko,A.Kozik,R.M.Michelmore,Z.Lai,L.H.Rieseberg,S.J.Knapp,SSRs and INDELs mined from the sunflower EST database:abundance,polymorphisms,and cross-taxa utility,Theor.Appl.Genet.117(2008)1021–1029.
[22]S.Tan,J.W.Cheng,L.Zhang,C.Qin,D.G.Nong,W.P.Li,X.Tang,Z.M.Wu,K.L.Hu,Construction of an interspecific genetic map based on InDel and SSR for mapping the QTLs affecting the initiation of flower primordia in pepper(Capsicum spp.),PLoS One 10(2015),e0119389..
[23]W.P.Li,J.W.Cheng,Z.M.Wu,C.Qin,S.Tan,X.Tang,J.J.Cui,L.Zhang,K.L.Hu,An InDel-based linkage map of hot pepper(Capsicum annuum),Mol.Breed.35(2015)32.
[24]K.Wu,M.M.Yang,H.Y.Liu,Y.Tao,J.Mei,Y.Z.Zhao,Genetic analysis and molecular characterization of Chinese sesame(Sesamum indicum L.)cultivars using insertion-deletion(InDel)and simple sequence repeat(SSR)markers,BMC Genet.15(2014)35.
[25]X.M.Li,W.H.Gao,H.L.Guo,X.L.Zhang,D.D.Fang,Z.X.Lin,Development of EST-based SNP and InDel markers and their utilization in tetraploid cotton genetic mapping,BMC Genomics 15(2014)1046.
[26]F.Ollitrault,J.Terol,A.A.Martin,J.A.Pina,L.Navarro,M.Talon,P.Ollitrault,Development of indel markers from Citrus clementina(Rutaceae)BAC-end sequences and interspecific transferability in Citrus,Am.J.Bot.99(2012)e268–e273.
[27]A.Garcia-Lor,F.Luro,L.Navarro,P.Ollitrault,Comparative use of InDel and SSR markers in deciphering the interspecific structure of cultivated citrus genetic diversity:a perspective for genetic association studies,Mol.Gen.Genomics.287(2012)77–94.
[28]H.B.Sohn,S.J.Kim,T.Y.Hwang,H.M.Park,Y.Y.Lee,K.Markkandan,D.Lee,S.Lee,S.Y.Hong,Y.H.Song,B.C.Koo,Y.H.Kim,Barcode system for genetic identification of soybean[Glycine max(L.)Merrill]cultivars using InDel markers specific to dense variation blocks,Front.Plant Sci.8(2017)520.
[29]X.F.Song,H.C.Wei,W.Cheng,S.X.Yang,Y.X.Zhao,X.Li,D.Luo,H.Zhang,X.Z.Feng,Development of INDEL markers for genetic mapping based on whole genome resequencing in soybean,G3-Genes Genomes Genet.5(2015)2793–2799.
[30]J.Schmutz,S.B.Cannon,J.Schlueter,J.X.Ma,T.Mitros,W.Nelson,D.L.Hyten,Q.J.Song,J.J.Thelen,J.L.Cheng,D.Xu,U.Hellsten,G.D.May,Y.Yu,T.Sakurai,T.Umezawa,M.K.Bhattacharyya,D.Sandhu,B.Valliyodan,E.Lindquist,M.Peto,D.Grant,S.Q.Shu,D.Goodstein,K.Barry,M.Futrell-Griggs,B.Abernathy,J.C.Du,Z.X.Tian,L.C.Zhu,N.Gill,T.Joshi,M.Libault,A.Sethuraman,X.C.Zhang,K.Shinozaki,H.T.Nguyen,R.A.Wing,P.Cregan,J.E.Specht,J.Grimwood,D.Rokhsar,G.Stacey,R.C.Shoemaker,S.A.Jackson,Genome sequence of the palaeopolyploid soybean,Nature 463(2010)178–183.
[31]M.Y.Kim,S.Lee,K.Van,T.H.Kim,S.C.Jeong,I.Y.Choi,D.S.Kim,Y.S.Lee,D.Park,J.Ma,W.Y.Kim,B.C.Kim,S.Park,K.A.Lee,D.H.Kim,K.H.Kim,J.H.Shin,Y.E.Jang,K.D.Kim,W.X.Liu,T.Chaisan,Y.J.Kang,Y.H.Lee,K.H.Kim,J.K.Moon,J.Schmutz,S.A.Jackson,J.Bhak,S.H.Lee,Whole-genome sequencing and intensive analysis of the undomesticated soybean(Glycine soja Sieb.and Zucc.)genome,Proc.Natl.Acad.Sci.U.S.A.107(2010)22032–22037.
[32]H.M.Lam,X.Xu,X.Liu,W.B.Chen,G.H.Yang,F.L.Wong,M.W.Li,W.M.He,N.Qin,B.Wang,J.Li,M.Jian,J.Wang,G.H.Shao,J.Wang,S.S.Sun,G.Y.Zhang,Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection,Nat.Genet.42(2010)1053–1059.
[33]Y.H.Kim,H.M.Park,T.Y.Hwang,S.K.Lee,M.S.Choi,S.Jho,S.Hwang,H.M.Kim,D.Lee,B.C.Kim,C.P.Hong,Y.S.Cho,H.Kim,K.H.Jeong,M.J.Seo,H.T.Yun,S.L.Kim,Y.U.Kwon,W.H.Kim,H.K.Chun,S.J.Lim,Y.A.Shin,I.Y.Choi,Y.S.Kim,H.S.Yoon,S.H.Lee,S.Lee,Variation block-based genomics method for crop plants,BMC Genomics 15(2014)477.
[34]W.H.Chung,N.Jeong,J.Kim,W.K.Lee,Y.G.Lee,S.H.Lee,W.Yoon,J.H.Kim,I.Y.Choi,H.K.Choi,J.K.Moon,N.Kim,S.C.Jeong,Population structure and domestication revealed by high-depth resequencing of Korean cultivated and wild soybean genomes,DNA Res.21(2014)153–167.
[35]Z.K.Zhou,Y.Jiang,Z.Wang,Z.H.Gou,J.Lyu,W.Y.Li,Y.J.Yu,L.P.Shu,Y.J.Zhao,Y.M.Ma,C.Fang,Y.T.Shen,T.F.Liu,C.C.Li,Q.Li,M.Wu,M.Wang,Y.S.Wu,Y.Dong,W.T.Wan,X.Wang,Z.L.Ding,Y.D.Gao,H.Xiang,B.G.Zhu,S.H.Lee,W.Wang,Z.X.Tian,Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean,Nat.Biotechnol.33(2015)408–414.
[36]M.Murray,W.F.Thompson,Rapid isolation of high molecular weight plant DNA,Nucleic Acids Res.8(1980)4321–4326.
[37]H.Li,R.Durbin,Fast and accurate short read alignment with Burrows-Wheeler transform,Bioinformatics 25(2009)1754–1760.
[38]H.Li,A statistical framework for SNP calling,mutation discovery,association mapping and population genetical parameter estimation from sequencing data,Bioinformatics 27(2011)2987–2993.
[39]A.McKenna,M.Hanna,E.Banks,A.Sivachenko,K.Cibulskis,A.Kernytsky,K.Garimella,D.Altshuler,S.Gabriel,M.Daly,M.A.DePristo,The Genome Analysis Toolkit:a MapReduce framework for analyzing next-generation DNA sequencing data,Genome Res.20(2010)1297–1303.
[40]D.C.Koboldt,K.Chen,T.Wylie,D.E.Larson,M.D.McLellan,E.R.Mardis,G.M.Weinstock,R.K.Wilson,L.Ding,VarScan:variant detection in massively parallel sequencing of individual and pooled samples,Bioinformatics 25(2009)2283–2285.
[41]K.Ye,M.H.Schulz,Q.Long,R.Apweiler,Z.M.Ning,Pindel:a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads,Bioinformatics 25(2009)2865–2871.
[42]Y.H.Li,S.C.Zhao,J.X.Ma,D.Li,L.Yan,J.Li,X.T.Qi,X.S.Guo,L.Zhang,W.M.He,R.Z.Chang,Q.S.Liang,Y.Guo,C.Ye,X.B.Wang,Y.Tao,R.X.Guan,J.Y.Wang,Y.L.Liu,L.G.Jin,X.Q.Zhang,Z.X.Liu,L.J.Zhang,J.Chen,K.J.Wang,R.Nielsen,R.Q.Li,P.Y.Chen,W.B.Li,J.C.Reif,M.Purugganan,J.Wang,M.C.Zhang,J.Wang,L.J.Qiu,Molecular footprints of domestication and improvement in soybean revealed by whole genome re-sequencing,BMC Genomics 14(2013)579.
[43]C.C.Chang,C.J.Lin,LIBSVM:a library for support vector machines,ACM Trans.Intell.Syst.Technol.2(2011)27.
[44]A.Untergasser,I.Cutcutache,T.Koressaar,J.Ye,B.C.Faircloth,M.Remm,S.G.Rozen,Primer3-new capabilities and interfaces,Nucleic Acids Res.40(2012),e115..
[45]J.W.Van Ooijen,JoinMap4.0,Software for the calculation of genetic linkage maps in experimental populations,Kyazma B.V.,Wageningen,Netherlands,2006.
[46]H.H.Li,G.Y.Ye,J.K.Wang,A modified algorithm for the improvement of composite interval mapping,Genetics 175(2007)361–374.
[47]J.W.Van Ooijen,MapQTL 5,Software for the mapping of quantitative trait loci in experimental populations,Kyazma B.V.,Wageningen,Netherlands,2004.
[48]O.Henegariu,N.A.Heerema,S.R.Dlouhy,G.H.Vance,P.H.Vogt,P.C.R.Multiplex,Critical parameters and step-by-step protocol,BioTechniques 23(1997)504–511.
[49]E.R.Cober,S.J.Molnar,M.Charette,H.D.Voldeng,A new locus for early maturity in soybean,Crop Sci.50(2010)524–527.
[50]L.R.Cheng,Y.Wang,C.B.Zhang,C.X.Wu,J.L.Xu,H.Y.Zhu,J.T.Leng,Y.N.Bai,R.X.Guan,W.S.Hou,L.J.Zhang,T.F.Han,Genetic analysis and QTL detection of reproductive period and post-flowering photoperiod responses in soybean,Theor.Appl.Genet.123(2011)421–429.
[51]M.L.Xu,N.Yamagishi,C.Zhao,R.Takeshima,M.Kasai,S.Watanabe,A.Kanazawa,N.Yoshikawa,B.H.Liu,T.Yamada,J.Abe,The soybean-specific maturity gene E1 family of floral repressors controls night-break responses through downregulation of FLOWERING LOCUS T orthologs,Plant Physiol.168(2015)1735–1746.
猜你喜欢
现代青年·精英版(2021年9期)2021-10-08
家庭科学·新健康(2021年5期)2021-06-21
当代水产(2019年5期)2019-07-25
妇女生活(2019年7期)2019-07-16
疯狂英语·新悦读(2019年5期)2019-05-15
中国诗歌(2018年5期)2018-11-14
人大建设(2018年1期)2018-04-18
饮食与健康·下旬刊(2016年9期)2016-05-14
祝您健康(2015年6期)2015-09-16
环球时报(2012-02-10)2012-02-10

- The Crop Journal的其它文章
- Integrated physiological and molecular approaches to improvement of abiotic stress tolerance in two pulse crops of the semi-arid tropics
- Genome-wide association study of heat stresstolerance traits in spring-type Brassica napus L.under controlled conditions
- Zinc partitioning in basmati rice varieties as influenced by Zn fertilization
- Elevated temperature intensity, timing, and duration of exposure affect soybean internode elongation, mainstem node number, and pod number per plant
- Soybean hairy roots produced in vitro by Agrobacterium rhizogenes-mediated transformation
- Development and validation of simple sequence repeat markers from Arachis hypogaea transcript sequences