
Development and validation of simple sequence repeat markers from Arachis hypogaea transcript sequences
2018-04-12 03:33:55HoumioWngYongLeiLiyingYnLiyunWnYnCiZefengYngJinweiLvXiojieZhngChenwuXuBoshouLio
The Crop Journal 2018年2期
Houmio Wng,Yong Lei,Liying Yn,Liyun Wn,Yn Ci,Zefeng Yng,Jinwei Lv,Xiojie Zhng,Chenwu Xu,*,Boshou Lio,*
a Key Laboratory of Oil Crop Biology of the Ministry of Agriculture,CAAS-ICRISAT Joint Laboratory for Groundnut Aflatoxin Management,Oil Crops Research Institute of Chinese Academy of Agricultural Sciences,Wuhan 430062,Hubei,China
b Jiangsu Key Laboratory of Crop Genetics and Physiology/Co-Innovation Center for Modern Production Technology of Grain Crops,Key Laboratory of Plant Functional Genomics of the Ministry of Education,Yangzhou University,Yangzhou 225009,Zhejiang,China
1.Introduction
The cultivated peanut(Arachis hypogaea L.)is an important oilseed and cash crop in most tropical and subtropical areas of the world,and one of the primary sources of vegetable oil and protein for human consumption.The species is a self-pollinating allotetraploid(AABB)with two different genomes(A and B),and the genome size is estimated to be 2.8 Gb[1].The most likely wild diploid progenitors of A.hypogaea are A.duranensis(AA,2n=2x=20)and A.ipaensis(BB,2n=2x=20)[2].Reference genomes of A.duranensis(A genome)and A.ipaensis(B genome)have been released recently in a public database(http://peanutbase.org/).However,a reference genome of A.hypogaea is not yet available.
Molecular markers are valuable tools for linkage map construction,quantitative trait locus(QTL)analyses,genomic selection,gene discovery,and marker-assisted selection for crop improvement[3].They are also useful for estimating diversity and discriminating among genotypes[4].Progress has been made in the development of molecular markers and genetic resources in A.hypogaea[3,5–7].However,the application of molecular markers is more advanced in the legume species Glycine max and Medicago truncatula than in A.hypogaea,primarily because of the genome complexity and the narrow genetic base of A.hypogaea.Simple sequence repeats(SSRs)and single-nucleotide polymorphisms(SNPs)are currently the standard DNA markers used for gene mapping and marker-assisted selection in many crops[8].SSR and SNP markers share similar advantages,as both are codominant,abundant throughout genomes,and highly polymorphic.However,SSRs are often multi-allelic,whereas most SNPs are biallelic.SSRs can be easily detected by polymerase chain reaction(PCR)and gel electrophoresis[9].SSRs have been widely applied in A.hypogaea for verification of cultivar identity,diversity studies[7,10–15],linkage map construction[16–18],and QTL analysis[19–23].SSRs are classified into genomic SSRs and expressed sequence tag-SSRs(EST-SSRs)depending on the origin of the sequences used for the initial identification of these markers.Genomic SSRs are not necessarily expected either to have genetic functions or to be closely linked to transcribed regions of the genome,whereas EST-SSRs are tightly linked with functional genes that may influence important agronomic characters.Because of these advantages,EST-SSRs have been developed and used in many plant species[3,8,24–31].Although a major disadvantage of EST-SSRs is sequence redundancy,resulting in multiple sets of markers at the same locus,the problem can be circumvented by assembling the ESTs and short reads of RNA transcripts into unigenes.With a large number of EST resources of A.hypogaea in public databases,it is advisable to fully exploit the EST-SSRs within these sequences.Since Arachis species SSRs were first reported in 1999,a total of 14,390 A.hypogaea SSRs have been deposited to date in the public database(Peanut Marker Database,http://marker.kazusa.or.jp/Peanut/).However,the number of SSR markers reported for A.hypogaea is still far fewer than that reported for Glycine max[32].
The application of next-generation sequencing technologies has efficiently and cost-effectively generated a massive amount of genetics sequence data.Additionally,new techniques have enabled whole-transcriptome sequencing[i.e.,RNA sequencing(RNA-seq)]and analysis of crops[33].RNA-seq is an effective approach for detecting functional genes and characterizing gene expression patterns and associated regulatory networks.This technique has been used successfully to analyze the transcriptome of A.hypogaea under different conditions[34–39].RNA-seq has also allowed the rapid identification of SSR loci derived from ESTs in many crops[3,8,24–31].
We previously reported the first study of the post-harvest A.hypogaea transcriptome using RNA-seq and de novo assembly via Illumina paired-end sequencing[40].The raw sequencing data from that study were deposited in the National Center for Biotechnology Information(NCBI)Sequence Read Archive database(SRP061959),and 128,725 unigenes of A.hypogaea were obtained[40].In this study,these 128,725 unigene sequences were used to detect SSRs for the large-scale development and characterization of SSR markers.
2.Materials and methods
2.1.Plant materials and DNA extraction
Twenty-four A.hypogaea varieties from 14 provinces in China were used for analyzing the polymorphism of SSR markers(Table S1).All 24 varieties were planted in the experimental greenhouses of the Oil Crops Research Institute of the Chinese Academy of Agricultural Sciences(CAAS-OCRI),Wuhan,China.Genomic DNA was extracted from fresh leaves of each variety following the hexadecyltrimethyl ammonium bromide(CTAB)method[41].The quality and integrity of the extracted DNA were evaluated by 1.0%agarose gel electrophoresis and the concentrations were determined with a Beckman DU-650 spectrophotometer(Beckman Coulter,Inc.,Brea,CA,USA).
2.2.Expressed sequence tag simple sequence repeat detection and primer design
SSRs present in the 128,725 unigenes were detected using the MIcroSAtellite program(MISA,http://pgrc.ipk-gatersleben.de/misa/misa.html)[31].The default criteria were based on the minimum number of repeats,which were set as follows:10 repeating units for mononucleotides,six repeating units for dinucleotides,and five repeating units for tri-,tetra-,pentaand hexanucleotides.The maximum distance between two SSRs was specified as 100 bases.Primer pairs specific for the flanking regions of potential SSRs were designed for each SSR locus using Primer3(http://primer3.sourceforge.net/releases.php)[25].Primers were designed based on the following criteria:1)GC content between 40%and 60%,2)primer length between 18 and 27 bp,3)melting temperature between 57°C and 63°C,and 4)expected PCR product sizes from 100 to 280 bp.
2.3.Functional classification of simple sequence repeatcontaining unigenes
All unigenes containing an SSR motif were classified into Clusters of Orthologous Groups(COG)categories according to the results of National Center for Biotechnology Information(NCBI)BLAST(version 2.2.28+)searches against amino acid sequences in the Eukaryotic Orthologous Groups(KOG)database with an E-value threshold of 10−3(http://www.ncbi.nlm.nih.gov/COG/)[42].To comprehensively characterize the biological functions and interactions of these SSR-containing unigenes,pathways were assigned based on the KEGG database[43]using BLASTX with an E-value threshold of 10−5.
2.4.Validation of simple sequence repeats
Two hundred and ten SSR markers(Table S2)were validated using 24 A.hypogaea varieties(Table S1).PCR reactions were performed as previously described[41].The PCR-amplified products were separated by nondenaturing 6.0%polyacrylamide gel electrophoresis and then visualized by silver staining as described by Ren et al.[7]and Zhou et al.[44].The fragment sizes of the PCR products were estimated by comparison with a 50-bp DNA ladder.
2.5.Genetic diversity analysis
The number of alleles,genetic diversity(expected heterozygosity,He)and polymorphic information content(PIC)were estimated for each SSR using PowerMarker version 3.25[44,45].A genetic similarity matrix based on the proportion of shared alleles among the 24 A.hypogaea varieties was generated with PowerMarker.An unrooted neighbor-joining tree based on the shared allele distances was constructed using MEGA 6[44]to reveal the genetic relationships among the 24 varieties.
3.Results
3.1.Development and characterization of simple sequence repeats mined from the A.hypogaea transcriptome
All 128,725 unigenes assembled de novo from the A.hypogaea transcriptome(NCBI Sequence Read Archive database,SRP061959)with a total length of 98.47 Mb were used to identify potential SSR loci using MISA.A total of 29,357 potential SSRs were identified in 22,806 unigenes,with 4883 unigenes containing more than one SSR locus(Table 1).The distribution density was one SSR locus per 3355 bp,and the number of repeat units ranged from one to six.The number of SSRs with each repeat motif varied widely.SSRs with mononucleotide repeat motifs were most abundant(19,065;64.94%),followed by tri-(5033;17.14%),di-(4927;16.78%),tetra-(303;1.03%),penta-(18;0.061%),and hexanucleotide(11;0.037%)repeat motifs(Table 1,Fig.S1).Additionally,1710 SSRs were present in compound forms(Table 1).The iteration number of repeat units in SSRs ranged from 4 to 22 and the occurrence frequencies of SSRs with different iteration numbers were unequal.The most common iteration number was 10(8467;28.84%),followed by 11(3959;13.49%),five(3634;12.38%)and six(3507;11.95%)(Table S3,Fig.S1).For the SSRs with>10 repeat units,mononucleotide repeat motifs were most abundant,accounting for 99.82%of the SSRs,whereas motifs with>16 repeats were rare(5.79%).The repetition of sequences also varied.Sixty-nine SSR motifs were identified,including two mono-,four di-,10 tri-,24 tetra-,18 penta-,and 11 hexanucleotide repeating units(Table S3).The dominant motif identified in the SSRs was A/T(18,358;62.54%),followed by AG/CT(2804;9.55%),AAG/CTT(1396;4.76%),AT/AT(1390;4.73%),AAT/ATT(1075;3.66%),ATC/ATG(725;2.47%)and AC/GT(720;2.45%).The remaining 62 motifs were relatively rare,accounting for only 9.84%of the total number of SSRs(Fig.1).
Comparisons with known sequences in Kyoto Encyclopedia of Genes and Genomes(KEGG)and Eukaryotic Orthologous Groups(KOG)databases were used to categorize the SSR-containing unigenes based on functions.A search using the KEGG Orthology(KO)database revealed 1883(8.26%)SSR-containing unigenes with significant matches.These unigenes were assigned to five main categories,including 32 subcategories and 252 KEGG pathways(Fig.2,Table S4).The majority of the SSR-containing unigenes were assigned to“carbohydrate metabolism”(479;25.44%), “signal transduction”(385;20.45%),“overview”(314;16.68%)and “amino acid metabolism”(305;16.20%).Additionally,4103 SSR-containing unigenes were annotated using the KOG database and assigned to 26 KOG functional categories(Fig.3,Table S4).Among the 26 KOG categories,“general function prediction only”(693;16.89%)was the largest group,followed by“posttranslational modification,protein turnover,chaperones”(477;11.63%),“signal transduction mechanisms”(339;8.26%)and “transcription”(257;6.26%).The SSR-containing unigenes were functionally classified and characterized to enable molecular marker development for studying genetic diversity of A.hypogaea in the future.

Table 1–Summary of EST-SSRs identified in transcriptome sequences.
3.2.Primer design and identification of new simple sequence repeats
A total of 56,451 PCR primer pairs specific for the unique sequences flanking 18,817 SSR loci in 15,739 unigenes were designed(Table S5).For each SSR locus,three alternative primer pairs were designed.The flanking sequences of the other 10,540 SSRs did not fulfill the primer design criteria,which permitted no suitable PCR primer pairs for them.The 11,785 mononucleotide SSRs(10,585 unigenes)were the most common ones for which primers were successfully designed,followed by 3076 trinucleotide SSRs(2868 unigenes),2786 dinucleotide SSRs(2690 unigenes),980 complex SSRs(962),173 tetranucleotide SSRs(173 unigenes),12 pentanucleotide SSRs(12 unigenes),and five hexanucleotide SSRs(five unigenes)(Table 2).In total,primers for 5514 SSRs with di-to hexanucleotide motifs were designed.
In the Peanut Marker Database(http://marker.kazusa.or.jp/Peanut/,2016-01-27),14,390 primer pairs of the publicly available SSRs in A.hypogaea were searched(Table S6).Mononucleotide and complex SSRs were excluded during the identification of new SSRs.All 14,390 publicly available SSR primer pair sequences were aligned to the 22,806 unigenes as paired-end sequences,and 4340 new SSRs in 4064 unigenes were obtained for further analysis(Table S7).The proportions of new SSRs were not evenly distributed.The largest fraction of identified new SSRs consisted of di-and trinucleotide repeats,which accounted for 96.59%(4192)of all new SSRs.The other three types of new SSRs were rare(148,3.41%).
The novel SSR motifs were grouped into classes I and II based on their length[15,46].Among the new SSRs,the length of 228(5.25%)SSR motifs were≥20 bp(class I),whereas the other 4112 consisted of<20 bp(class II)(Table 3,Table S5).Thus,the number of class II SSRs was much greater than that of class I,a result consistent with previous reports in A.hypogaea[15,46,47].The number of tetranucleotides(133)was greater than that of other repeat motifs in the class I SSRs,whereas the proportion of trinucleotide was higher than those of di-,hexa-,tetra-,and pentanucleotides in class II SSRs(Table 3).Interestingly,tetra-,penta-,and hexanucleotide repeat motifs were detected only in class I(Table 3).
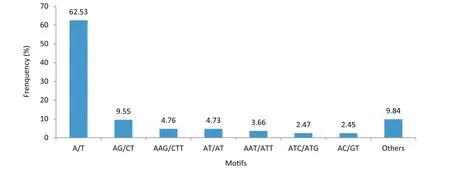
Fig.1–Frequency distribution of simple sequence repeats based on motif type.
3.3.Validation of novel simple sequence repeat markers
To validate the identified novel SSR markers,we attempted to amplify the predicted SSRs via PCR.A total of 210 primer pairs(Table S2)were randomly selected for validation using DNA from 24 A.hypogaea varieties(Table S1).The numbers of these selected SSRs with di-,tri-,and tetranucleotide repeats were 35,153,and 22,respectively.Among the 210 primer pairs,191(90.95%)were able to amplify genomic DNA and the containing SSRs with di-,tri-,and tetranucleotide repeats were 32(16.75%),140(73.30%),and 19(9.95%),respectively,whereas the remaining 19 primer pairs failed to generate PCR products at the same annealing temperatures(Table S2).Most of the selected markers appeared as single alleles in all 24 A.hypogaea genotypes,except for a few multilocus SSRs,suggesting that these novel SSR markers possess a specific amplification in A.hypogaea.
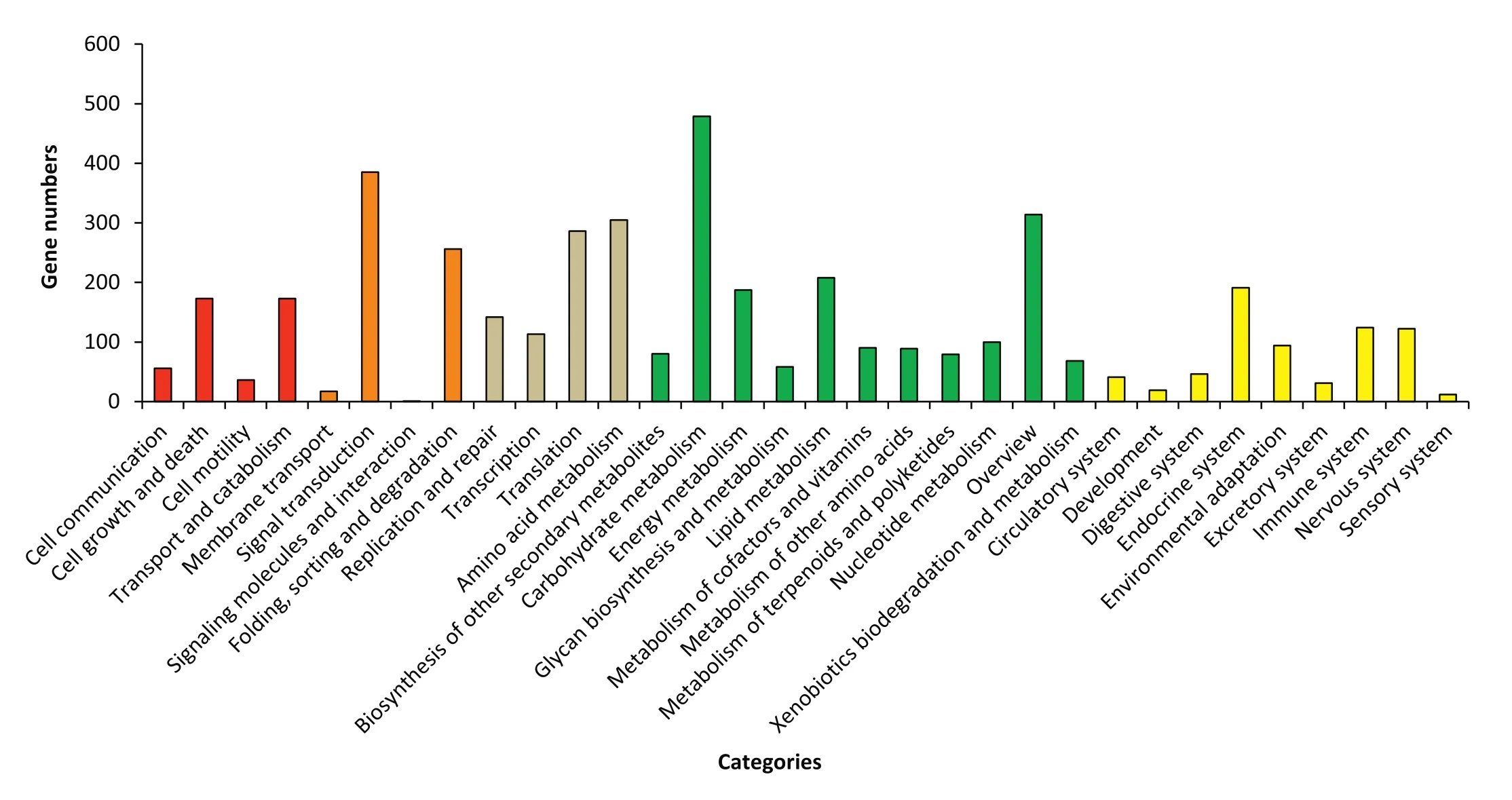
Fig.2–Functional classification of simple sequence repeat-containing unigenes based on Kyoto Encyclopedia of Genes and Genomes Ortholog searches.
Among the validated markers,37(17.62%)showed polymorphism between at least two A.hypogaea varieties,including new SSRs with di-(11;29.73%),tri-(25;67.57%),and tetranucleotide(1;2.70%)repeats(Table S2).Based on the polymorphism rate,about 765 polymorphic SSR markers were expected for the 4340 new SSRs.The 37 polymorphic SSR markers detected 146 alleles in total(N=24),with 2–10 alleles per locus(average,3.95 alleles per locus)(Table 4).Of the 37,25(67.57%)were observed in more than three A.hypogaea genotypes and four(10.81%)showed polymorphism among>10 A.hypogaea varieties.
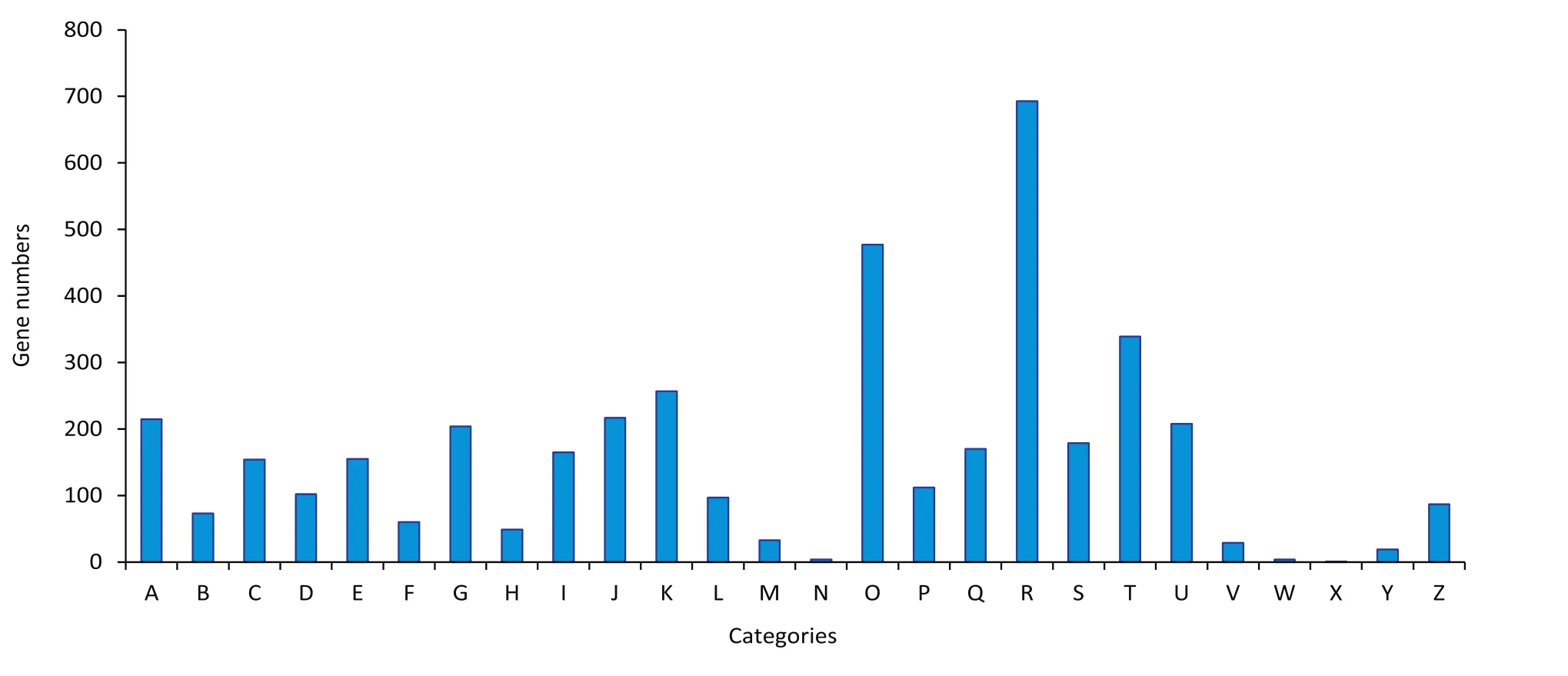
Fig.3–Functional classification of simple sequence repeat-containing unigenes based on the Eukaryotic Orthologous Groups of proteins database.A,RNA processing and modification;B,chromatin structure and dynamics;C,energy production and conversion;D,cell cycle control,cell division,chromosome partitioning;E,amino acid transport and metabolism;F,nucleotide transport and metabolism;G,carbohydrate transport and metabolism;H,coenzyme transport and metabolism;I,lipid transport and metabolism;J,translation,ribosomal structure and biogenesis;K,transcription;L,replication,recombination and repair;M,cell wall/membrane/envelope biogenesis;N,cell motility;O,posttranslational modification,protein turnover,chaperones;P,inorganic ion transport and metabolism;Q,secondary metabolite biosynthesis,transport and catabolism;R,general function prediction only;S,function unknown;T,signal transduction mechanisms;U,intracellular trafficking,secretion,and vesicular transport;V,defense mechanisms;W,extracellular structures;X,unnamed protein;Y,nuclear structure;Z,cytoskeleton.
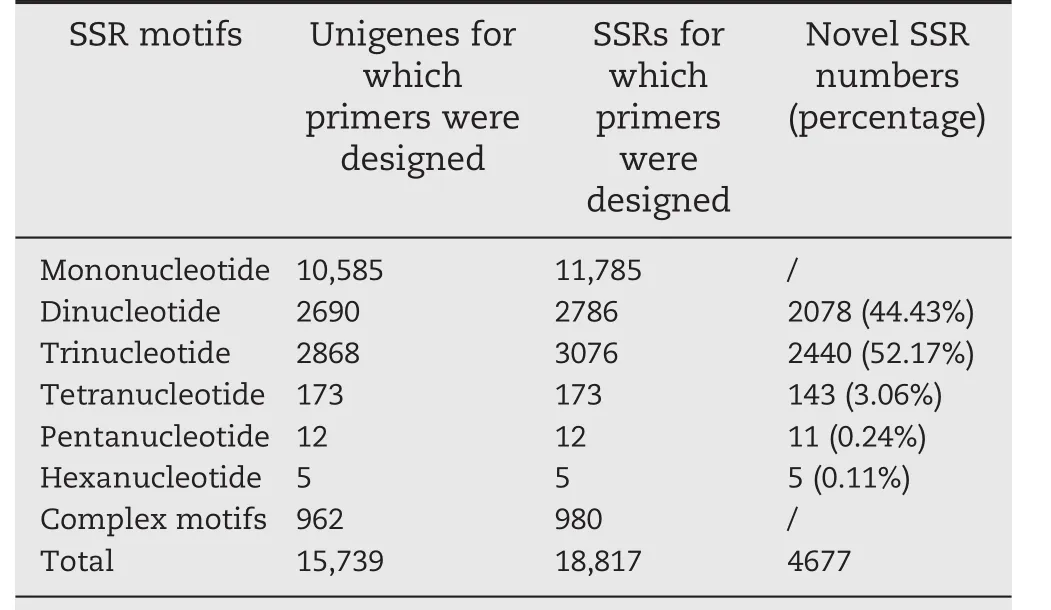
Table 2–Summary statistics of the simple sequence repeat(SSR)motifs designed with primers and distribution of new SSRs.
3.4.Evaluation of genetic diversity among 24 A.hypogaea varieties
The 37 polymorphic SSRs developed in this study were used to assess the genetic diversity and relationships among 24 A.hypogaea varieties cultivated across the A.hypogaea growing region in China (Table S1).The expectedheterozygosity(He)was 0.449(0.0799 to 0.8370)(Table 4),and the average polymorphic information content(PIC)value was 0.403(0.077 to 0.819)(Table 4).

Table 3–Classification of novel simple sequence repeats based on their motif lengths.
An unrooted neighbor-joining tree based on shared allele distance grouped the 24 A.hypogaea varieties into four main clusters(Fig.S2).The largest cluster included 13 genotypes,with eight(61.54%)from var.vulgaris(Table S1,Fig.S2).A few discrepancies were observed during the neighbor-joining tree analysis because of the relatively small number of validated polymorphic SSRs.Nevertheless,our results might suggest an association between genetic relationships of A.hypogaea varieties and their botanical species.
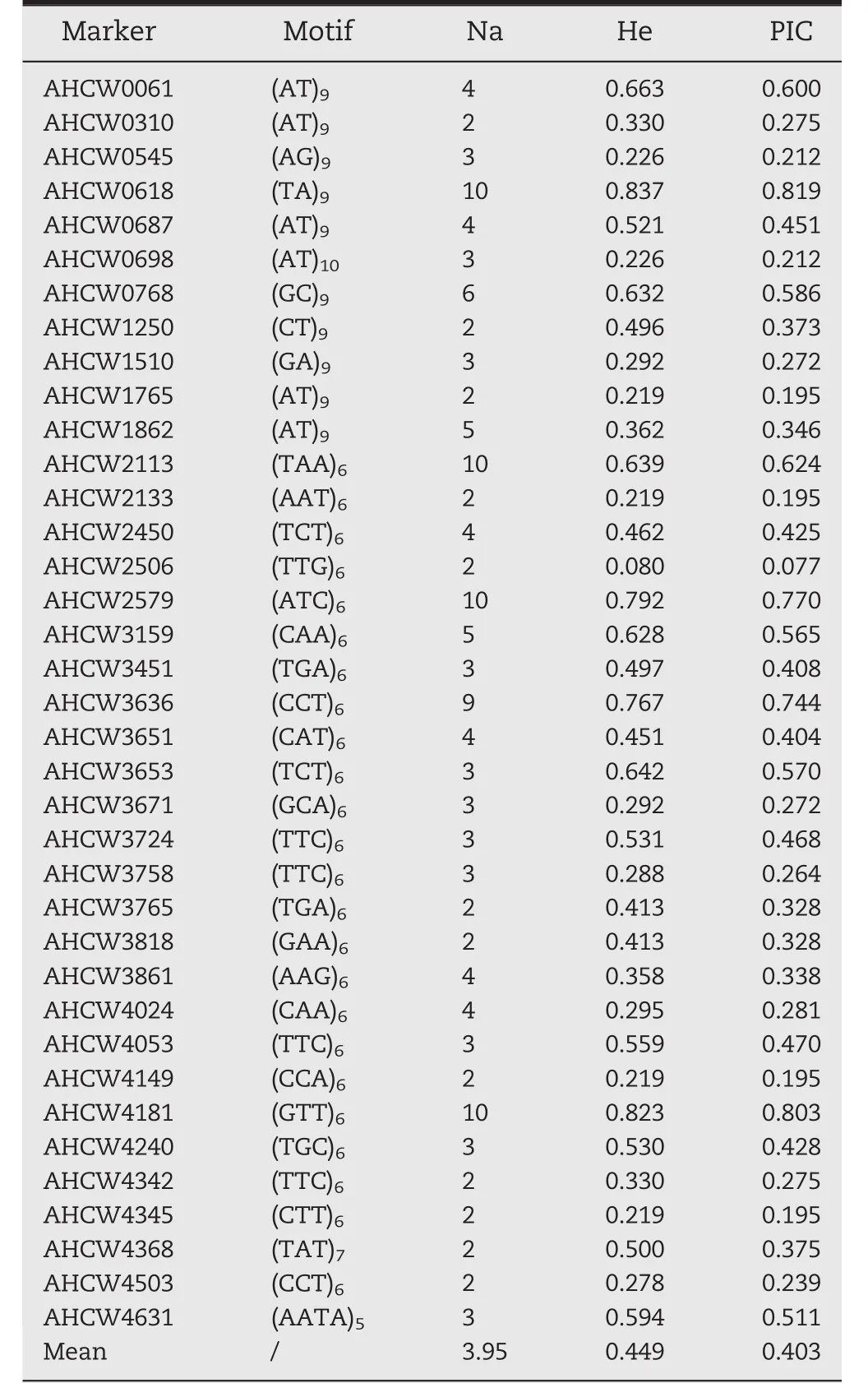
Table 4–Characteristics of 37 polymorphic simple sequence repeat markers in 24 peanut genotypes.
4.Discussion
The development of polymorphic genetic markers is an important task in studying the genetic basis of agronomic traits and population genetic structures, molecular marker-assisted selection,and QTL analysis.SSR markers are among the most useful molecular markers and have been applied for evaluating A.hypogaea genetic diversity and constructing several A.hypogaea genetic maps[3,7,41].Expressed sequence tags(ESTs)have become an important resource for developing SSR markers that are associated with biological function[3,8,24–31].In this study,we identified 29,357 potential SSRs based on the previously assembled A.hypogaea unigenes from our transcriptome sequencing study[40].Additionally,we detected 4340 new SSRs and designed corresponding primer pairs.Of the unigenes,17.7%contained SSRs,a proportion slightly higher than those reported in previous studies involving Arachis species(6.80%–16.95%)[3,15,36,48,49].Our observed proportion is also consistent with that reported for 49 dicotyledonous species(2.65%–10.62%)[50].The detection of SSRs is affected by several factors,including genome structure[50],dataset size for unigene assembly,and the criteria used for SSR mining[9].
The relative frequencies of di-,tri-,tetra-,penta-,and hexanucleotide motifs should decrease according to the relative probabilities of replication slippage events[51].The mononucleotide repeat motif was the most common among the analyzed A.hypogaea unigenes in this study.However,most studies have excluded mononucleotide repeat motifs because they may result from sequencing errors[52],and their polymorphism rate is very low.We observed similar proportions of di-and trinucleotide motifs in A.hypogaea transcript sequences,and these motifs were much more abundant than the tetra-,penta-,or hexanucleotide repeats.As previously reported[9,52],di-and trinucleotide motifs are generally most common in the SSRs of both dicotyledons and monocotyledons.The abundance of trinucleotide motifs observed in our study was consistent with the results of previous studies involving A.hypogaea[15,36,49,53]and other legumes[18,51,54,55].Trinucleotide repeat motifs are common for SSRs,as insertions or deletions within translated regions do not disturb open reading frames,whereas frameshift mutation may limit the expansion of other motif types[8,56].
In addition to the mononucleotide repeat motifs(19,065),>57.83%of the other 10,292 identified SSRs were matched with sequences in the Peanut Marker Database.These results suggest that our method is highly reliable for SSR development.Correspondingly,we identified 4340 new SSRs,which might be useful for constructing high-density genetic linkage maps,mapping QTL and using in crop breeding along with previously discovered molecular markers.The 91%validation rate observed in this study is consistent with the 85%–94%amplification rates of previous reports[3,15,44,46],indicating that the SSRs from the A.hypogaea transcriptomic data from high-throughput RNA-seq were suitable for SSR primer design.
Of the new SSRs,18(37)revealed polymorphism among the 24 A.hypogaea cultivars.These 37 polymorphic SSRs generated 146 alleles,and their PIC values varied from 0.077 to 0.819 with an average of 0.403(Table 4).The PIC value is used mainly to assess the utility of a marker for linkage analysis.The average PIC value(0.403)of the 37 polymorphic SSRs in our study was higher than those in two previous reports in A.hypogaea [15,44],suggesting that these informative markers may play vital roles in accelerating molecular genetics,marker-assisted selection breeding,germplasm polymorphism assessment,and functional genetics studies in A.hypogaea.
5.Conclusions
A total of 4030 novel SSR markers were identified and characterized in genomic data of A.hypogaea using high-throughput transcriptome sequencing.A set of 210 novel markers were validated in 24 A.hypogaea varieties.Of these,191(90.95%)yielded PCR products,and 37 polymorphic markers were identified among the 24 varieties.These new SSRs developed in this study will expand the current marker resources of A.hypogaea and may also be useful for functional genomics research and molecular breeding in A.hypogaea.
Supplementary data for this article can be found online at https://doi.org/10.1016/j.cj.2017.09.007.
This research was funded by the National Basic Research Program of China(2013CB127803,2011CB109304),National High Technology Research and Development Program of China(2013AA102602),National Natural Science Foundation of China (31371662,31461143022),China Agriculture Research System(CARS-14),and Shandong Agricultural Industrialization Project for New Variety Development(2014–2016).
[1]S.Feng,X.Wang,X.Zhang,P.M.Dang,C.C.Holbrook,A.K.Culbreath,Y.Wu,B.Guo,Peanut(Arachis hypogaea)expressed sequence tag project:progress and application,Comp.Funct.Genomics 2012(2012)373768.
[2]G.Kochert,H.T.Stalker,M.Gimenes,L.Galgaro,C.R.Lopes,K.Moore,RFLP and cytogenetic evidence on the origin and evolution of allotetraploid domesticated peanut,Arachis hypogaea(Leguminosae),Am.J.Bot.83(1996)1282–1291.
[3]Z.Peng,M.Gallo,B.L.Tillman,D.Rowland,J.Wang,Molecular marker development from transcript sequences and germplasm evaluation for cultivated peanut(Arachis hypogaea L.),Mol.Gen.Genomics.291(2016)363–381.
[4]M.Kirst,C.M.Cordeiro,G.Rezende,D.Grattapaglia,Power of microsatellite markers for fingerprinting and parentage analysis in Eucalyptus grandis breeding populations,J.Hered.96(2005)161–166.
[5]T.Iwashina,Flavonoid function and activity to plants and other organisms,Biol.Sci.Space 17(2003)24–44.
[6]R.K.Varshney,S.M.Mohan,P.M.Gaur,N.V.P.R.Gangarao,M.K.Pandey,A.Bohra,S.L.Sawargaonkar,A.Chitikineni,P.K.Kimurto,P.Janila,K.B.Saxena,A.Fikre,M.Sharma,A.Rathore,A.Pratap,S.Tripathi,S.Datta,S.K.Chaturvedi,N.Mallikarjuna,G.Anuradha,A.Babbar,A.K.Choudhary,M.B.Mhase,C.Bharadwaj,D.M.Mannur,P.N.Harer,B.Z.Guo,X.Q.Liang,N.Nadarajan,C.L.L.Gowda,Achievements and prospects of genomics-assisted breeding in three legume crops of the semi-arid tropics,Biotechnol.Adv.31(2013)1120–1134.
[7]X.P.Ren,H.F.Jiang,Z.Y.Yan,Y.N.Chen,X.J.Zhou,L.Huang,Y.Lei,J.Q.Huang,L.Y.Yan,Y.Qi,W.H.Wei,B.S.Liao,Genetic diversity and population structure of the major peanut(Arachis hypogaea L.)cultivars grown in China by SSR markers,PLoS One 9(2014)e0088091.
[8]H.Chen,L.Liu,L.Wang,S.Wang,P.Somta,X.Cheng,Development and validation of EST-SSR markers from the transcriptome of adzuki bean(Vigna angularis),PLoS One 10(2015),e0131939..
[9]R.K.Varshney,A.Graner,M.E.Sorrells,Genic microsatellite markers in plants:features and applications,Trends Biotechnol.23(2005)48–55.
[10]M.E.Ferguson,M.D.Burow,S.R.Schulze,P.J.Bramel,A.H.Paterson,S.Kresovich,S.Mitchell,Microsatellite identification and characterization in peanut(A-hypogaea L.),Theor.Appl.Genet.108(2004)1064–1070.
[11]M.S.Hopkins,A.M.Casa,T.Wang,S.E.Mitchell,R.E.Dean,G.D.Kochert,S.Kresovich,Discovery and characterization of polymorphic simple sequence repeats(SSRs)in peanut,Crop Sci.39(1999)1243–1247.
[12]Md.C.Moretzsohn,M.S.Hopkins,S.E.Mitchell,S.Kresovich,J.F.M.Valls,M.E.Ferreira,Genetic diversity of peanut(Arachis hypogaea L.)and its wild relatives based on the analysis of hypervariable regions of the genome,BMC Plant Biol.4(2004)11.
[13]G.H.He,R.H.Meng,H.Gao,B.Z.Guo,G.Q.Gao,M.Newman,R.N.Pittman,C.S.Prakash,Simple sequence repeat markers for botanical varieties of cultivated peanut(Arachis hypogaea L.),Euphytica 142(2005)131–136.
[14]R.Tang,G.Gao,L.He,Z.Han,S.Shan,R.Zhong,C.Zhou,J.Jiang,Y.Li,W.Zhuang,Genetic diversity in cultivated groundnut based on SSR markers,J.Genet.Genomics 34(2007)449–459.
[15]T.C.Bosamia,G.P.Mishra,R.Thankappan,J.R.Dobaria,Novel and stress relevant EST derived SSR markers developed and validated in peanut,PLoS One 10(2015),e0129127..
[16]Y.Hong,X.Chen,X.Liang,H.Liu,G.Zhou,S.Li,S.Wen,C.C.Holbrook,B.Z.Guo,A SSR-based composite genetic linkage map for the cultivated peanut(Arachis hypogaea L.)genome,BMC Plant Biol.10(2010)17.
[17]H.Qin,S.Feng,C.Chen,Y.Guo,S.Knapp,A.Culbreath,G.H.He,M.L.Wang,X.Y.Zhang,C.C.Holbrook,P.Ozias-Akins,B.Z.Guo,An integrated genetic linkage map of cultivated peanut(Arachis hypogaea L.)constructed from two RIL populations,Theor.Appl.Genet.124(2012)653–664.
[18]K.Shirasawa,P.Koilkonda,K.Aoki,H.Hirakawa,S.Tabata,M.Watanabe,M.Hasegawa,H.Kiyoshima,S.Suzuki,C.Kuwata,Y.Naito,T.Kuboyama,A.Nakaya,S.Sasamoto,A.Watanabe,M.Kato,K.Kawashima,Y.Kishida,M.Kohara,A.Kurabayashi,C.Takahashi,H.Tsuruoka,T.Wada,S.Isobe,In silico polymorphism analysis for the development of simple sequence repeat and transposon markers and construction of linkage map in cultivated peanut,BMC Plant Biol.12(2012)80.
[19]Y.P.Khedikar,M.V.C.Gowda,C.Sarvamangala,K.V.Patgar,H.D.Upadhyaya,R.K.Varshney,A QTL study on late leaf spot and rust revealed one major QTL for molecular breeding for rust resistance in groundnut(Arachis hypogaea L.),Theor.Appl.Genet.121(2010)971–984.
[20]K.Ravi,V.Vadez,S.Isobe,R.R.Mir,Y.Guo,S.N.Nigam,M.V.C.Gowda,T.Radhakrishnan,D.J.Bertioli,S.J.Knapp,R.K.Varshney,Identification of several small main-effect QTLs and a large number of epistatic QTLs for drought tolerance related traits in groundnut(Arachis hypogaea L.),Theor.Appl.Genet.122(2011)1119–1132.
[21]I.Faye,M.K.Pandey,F.Hamidou,A.Rathore,O.Ndoye,V.Vadez,R.K.Varshney,Identification of quantitative trait loci for yield and yield related traits in groundnut(Arachis hypogaea L.)under different water regimes in Niger and Senegal,Euphytica 206(2015)631–647.
[22]L.Huang,H.Y.He,W.G.Chen,X.P.Ren,Y.N.Chen,X.J.Zhou,Y.L.Xia,X.L.Wang,X.G.Jiang,B.S.Liao,H.F.Jiang,Quantitative trait locus analysis of agronomic and quality-related traits in cultivated peanut(Arachis hypogaea L.),Theor.Appl.Genet.128(2015)1103–1115.
[23]M.L.Wang,P.Khera,M.K.Pandey,H.Wang,L.Qiao,S.Feng,B.Tonnis,N.A.Barkley,D.Pinnow,C.C.Holbrook,A.K.Culbreath,R.K.Varshney,B.Z.Guo,Genetic mapping of QTLs controlling fatty acids provided insights into the genetic control of fatty acid synthesis pathway in peanut(Arachis hypogaea L.),PLoS One 10(2015)e0119454.
[24]T.Liu,S.Zhu,L.Fu,Q.Tang,Y.Yu,P.Chen,M.Luan,C.Wang,S.Tang,Development and characterization of 1,827 expressed sequence tag-derived simple sequence repeat markers for ramie(Boehmeria nivea L.Gaud),PLoS One 8(2013)e60346.
[25]W.Chen,Y.X.Liu,G.F.Jiang,De novo assembly and characterization of the testis transcriptome and development of EST-SSR markers in the cockroach Periplaneta americana,Sci.Rep.5(2015)11144.
[26]Q.Ding,J.Li,F.Wang,Y.Zhang,H.Li,J.Zhang,J.Gao,Characterization and development of EST-SSRs by deep transcriptome sequencing in Chinese cabbage(Brassica rapa L.ssp.pekinensis),Int.J.Genomics 473028(2015).
[27]Y.F.Guo,K.E.Wiegert-Rininger,V.A.Vallejo,C.S.Barry,R.M.Warner,Transcriptome-enabled marker discovery and mapping of plastochron-related genes in Petunia spp.BMC Genomics 16(2015)726.
[28]Y.Liu,P.Zhang,M.Song,J.Hou,M.Qing,W.Wang,C.Liu,Transcriptome analysis and development of SSR molecular markers in Glycyrrhiza uralensis fisch,PLoS One 10(2015),e0143017.
[29]C.Luo,H.X.Wu,Q.S.Yao,S.B.Wang,W.T.Xu,Development of EST-SSR and TRAP markers from transcriptome sequencing data of the mango,Genet.Mol.Res.14(2015)7914–7919.
[30]S.Y.Zhang,C.Feng,C.J.Xu,C.Q.Zhu,K.S.Chen,Polymorphisms in different EST-SSR types derived from the Chinese bayberry(Myrica rubra,Myricaceae)transcriptome,Genet.Mol.Res.14(2015)6037–6041.
[31]X.J.Zhou,Y.Y.Wang,Y.N.Xu,R.S.Yan,P.Zhao,W.Z.Liu,De novo characterization of flower bud transcriptomes and the development of EST-SSR markers for the endangered tree Tapiscia sinensis,Int.J.Mol.Sci.16(2015)12855–12870.
[32]D.Xin,J.Sun,J.Wang,H.Jiang,G.Hu,C.Liu,Q.Chen,Identification and characterization of SSRs from soybean(Glycine max)ESTs,Mol.Biol.Rep.39(2012)9047–9057.
[33]S.Li,S.W.Tighe,C.M.Nicolet,D.Grove,S.Levy,W.Farmerie,A.Viale,C.Wright,P.A.Schweitzer,Y.Gao,D.Kim,J.Boland,B.Hicks,R.Kim,S.Chhangawala,N.Jafari,N.Raghavachari,J.Gandara,N.Garcia-Reyero,C.Hendrickson,D.Roberson,J.Rosenfeldr,T.Smith,J.G.Underwood,M.Wang,P.Zumbo,D.A.Baldwin,G.S.Grills,C.E.Mason,Multi-platform assessment of transcriptome profiling using RNA-seq in the ABRF next-generation sequencing study,Nat.Biotechnol.32(2014)915–925.
[34]Y.N.Chen,X.P.Ren,X.J.Zhou,L.Huang,L.Y.Yan,Y.Lei,B.S.Liao,H.F.Jiang,Dynamics in the resistant and susceptible peanut(Arachis hypogaea L.)root transcriptome on infection with the Ralstonia solanacearum,BMC Genomics 15(2014)1078.
[35]L.Geng,X.Duan,C.Liang,C.Shu,F.Song,J.Zhang,Mining tissue-specific contigs from peanut(Arachis hypogaea L.)for promoter cloning by deep transcriptome sequencing,Plant Cell Physiol.55(2014)1793–1801.
[36]P.M.Guimaraes,A.C.M.Brasileiro,C.V.Morgante,A.C.Q.Martins,G.Pappas,O.B.Silva Jr.,R.Togawa,S.C.M.Leal-Bertioli,A.C.G.Araujo,M.C.Moretzsohn,D.J.Bertioli,Global transcriptome analysis of two wild relatives of peanut under drought and fungi infection,BMC Genomics 13(2012)387.
[37]X.Li,J.Lu,S.Liu,X.Liu,Y.Lin,L.Li,Identification of rapidly induced genes in the response of peanut(Arachis hypogaea)to water deficit and abscisic acid,BMC Biotechnol.14(2014)58.
[38]D.Yin,Y.Wang,X.Zhang,H.Li,X.Lu,J.Zhang,W.Zhang,S.Chen,De novo assembly of the peanut(Arachis hypogaea L.)seed transcriptome revealed candidate unigenes for oil accumulation pathways,PLoS One 8(2013)e73767.
[39]W.Zhu,X.P.Chen,H.Li,F.Zhu,Y.B.Hong,R.K.Varshney,X.Q.Liang,Comparative transcriptome analysis of aerial and subterranean pods development provides insights into seed abortion in peanut,Plant Mol.Biol.85(2014)395–409.
[40]H.M.Wang,Y.Lei,L.Y.Wan,L.Y.Yan,J.W.Lv,X.F.Dai,X.P.Ren,W.Guo,H.F.Jiang,B.S.Liao,Comparative transcript profiling of resistant and susceptible peanut post-harvest seeds in response to aflatoxin production by Aspergillus flavus,BMC Plant Biol.16(2016)54.
[41]L.M.Cuc,E.S.Mace,J.H.Crouch,V.D.Quang,T.D.Long,R.K.Varshney,Isolation and characterization of novel microsatellite markers and their application for diversity assessment in cultivated groundnut(Arachis hypogaea),BMC Plant Biol.8(2008)55.
[42]R.L.Tatusov,N.D.Fedorova,J.D.Jackson,A.R.Jacobs,B.Kiryutin,E.V.Koonin,D.M.Krylov,R.Mazumder,S.L.Mekhedov,A.N.Nikolskaya,B.S.Rao,S.Smirnov,A.V.Sverdlov,S.Vasudevan,Y.I.Wolf,J.J.Yin,D.A.Natale,The COG database:an updated version includes eukaryotes,BMC Bioinf.4(2003)41.
[43]M.Kanehisa,S.Goto,KEGG:Kyoto encyclopedia of genes and genomes,Nucleic Acids Res.28(2000)27–30.
[44]I.Ahuja,R.Kissen,A.M.Bones,Phytoalexins in defense against pathogens,Trends Plant Sci.17(2012)73–90.
[45]K.J.Liu,S.V.Muse,PowerMarker:an integrated analysis environment for genetic marker analysis,Bioinformatics 21(2005)2128–2129.
[46]V.Arbona,A.Gomez-Cadenas,Metabolomics of disease resistance in crops,Curr.Issues Mol.Biol.19(2016)13–29.
[47]S.S.Arya,A.R.Salve,S.Chauhan,Peanuts as functional food:a review,J.Food Sci.Technol.-Mysore 53(2016)31–41.
[48]X.Q.Liang,X.P.Chen,Y.B.Hong,H.Y.Liu,G.Y.Zhou,S.X.Li,B.Z.Guo,Utility of EST-derived SSR in cultivated peanut(Arachis hypogaea L.)and Arachis wild species,BMC Plant Biol.9(2009)35.
[49]P.Koilkonda,S.Sato,S.Tabata,K.Shirasawa,H.Hirakawa,H.Sakai,S.Sasamoto,A.Watanabe,T.Wada,Y.Kishida,H.Tsuruoka,T.Fujishiro,M.Yamada,M.Kohara,S.Suzuki,M.Hasegawa,H.Kiyoshima,S.Isobe,Large-scale development of expressed sequence tag-derived simple sequence repeat markers and diversity analysis in Arachis spp.Mol.Breed.30(2012)125–138.
[50]S.P.Kumpatla,S.Mukhopadhyay,Mining and survey of simple sequence repeats in expressed sequence tags of dicotyledonous species,Genome 48(2005)985–998.
[51]S.Kaur,L.W.Pembleton,N.O.Cogan,K.W.Savin,T.Leonforte,J.Paull,M.Materne,J.W.Forster,Transcriptome sequencing of field pea and faba bean for discovery and validation of SSR genetic markers,BMC Genomics 13(2012)104.
[52]Y.M.Zhao,T.Zhou,Z.H.Li,G.F.Zhao,Characterization of global transcriptome using Illumina paired-end sequencing and development of EST-SSR markers in two species of gynostemma(Cucurbitaceae),Molecules 20(2015)21214–21231.
[53]G.Q.Song,M.J.Li,H.Xiao,X.J.Wang,R.H.Tang,H.Xia,C.Z.Zhao,Y.P.Bi,EST sequencing and SSR marker development from cultivated peanut(Arachis hypogaea L.),Electron.J.Biotechnol.13(2010)1010.
[54]G.Agarwal,S.Jhanwar,P.Priya,V.K.Singh,M.S.Saxena,S.K.Parida,R.Garg,A.K.Tyagi,M.Jain,Comparative analysis of Kabuli chickpea transcriptome with desi and wild chickpea provides a rich resource for development of functional markers,PLoS One 7(2012),e52443.
[55]Z.Wang,G.Yu,B.Shi,X.Wang,H.Qiang,H.Gao,Development and characterization of simple sequence repeat(SSR)markers based on RNA-sequencing of Medicago sativa and in silico mapping onto the M.truncatula genome,PLoS One 9(2014)e92029.
[56]D.Metzgar,J.Bytof,C.Wills,Selection against frameshift mutations limits microsatellite expansion in coding DNA,Genome Res.10(2000)72–80.

- The Crop Journal的其它文章
- Genetic diversity assessment of a set of introduced mung bean accessions(Vigna radiata L.)
- Near-infrared reflectance spectroscopy reveals wide variation in major components of sesame seeds from Africa and Asia
- Alternate phenotype–genotype selection for developing superior high-yielding irrigated rice lines
- F unction of the auxin-responsive gene TaSAUR75 under salt and drought stress
- Soybean hairy roots produced in vitro by Agrobacterium rhizogenes-mediated transformation
- Elevated temperature intensity, timing, and duration of exposure affect soybean internode elongation, mainstem node number, and pod number per plant