
连枷臂综合征3例报告及文献复习
2018-03-13 02:23解婷婷张海宁冯加纯
中风与神经疾病杂志 2018年1期
王 爽, 李 超, 解婷婷, 张海宁, 冯加纯
连枷臂综合征(flail arm syndrome,FAS)是肌萎缩侧索硬化(amyotrophic lateral sclerosis,ALS)的变异型,以对称性双上肢近端肌无力和肌萎缩为主要特征,而双下肢、球部肌肉仅轻微受累,预后较ALS好,生存期也相对较长。由于其进展缓慢,在疾病早期易误诊、漏诊[1]。现将2015年~2016年我院收治的3例FAS病例进行分析并结合文献总结其临床特点。
1 病例资料
3例患者均为男性;年龄59岁~77岁,平均68岁;均为隐匿起病,主要表现为双上肢无力,2例以右上肢(优势侧肢体)无力起病,1例以双上肢无力起病;1例有高血压病史及第4腰椎骨折史,余2例既往体健;3例均有不同程度的上肢近端肌肉萎缩,2例可见肌肉颤动,3例上肢腱反射均消失,2例上肢肌张力下降,1例上肢可见病理反射,3例均无球部及双下肢体征;3例肌电图均有神经源性损害,1例行肌肉活检显示神经源性病理改变,2例未做肌肉活检,2例肌酸激酶轻中度升高;2例患者1 y后随访1例病情未见明显变化,1例双上肢无力加重。3例患者入院后肺部CT、腹部彩超、血常规、肝肾功、甲功、离子、肿瘤标志物、风湿免疫系列、外科综合等均未见异常。临床资料汇总见表1。
典型病例:病例1 男性,59岁,因“双上肢无力进行性加重3 y”入院。患者3 y前无明显诱因出现右上肢无力,尚可平举,1 y前累及左上肢,双上肢无力呈进行性加重,表现为不能抬起及持物,伴双手肿胀及麻木,自述双上肢偶有“肉跳感”,无饮水呛咳、吞咽困难、双下肢活动不灵。否认家族史。神经系统查体:神清语明,脑神经(-),双侧岗上肌、岗下肌、三角肌、肱二头肌、肱三头肌可见明显萎缩。深、浅感觉未见明显异常。双上肢肌力0级,双下肢肌力5级。双上肢肌张力减退,双下肢肌张力正常。跟膝胫试验稳准。双上肢腱反射消失,双下肢腱反射对称引出。双侧病理征阴性,无项强,克氏征阴性。实验室检查:肌电图(2014年8月,中国医科大学附属盛京医院):(1)双上肢所检肌广泛神经源性损害(均可见失神经电位,MUP时限增宽);(2)右侧正中神经、尺神经运动传导M波低,远端潜伏期延长,SCV无明显异常;(3)右侧胸锁乳突肌、左侧颈前肌MUP未见异常。左侧肱二头肌肌肉活检(2015年7月,北京大学第一临床医学院):所检骨骼肌呈神经源性病理改变。肌电图(2016年10月,吉林大学第一医院):双上肢肌及胸锁乳突肌呈神经源性损害,双下肢肌未见明显异常。头部MRI及颈椎MRI:未见明显异常。肌酸激酶 523 U/L (25~200)。诊断:FAS。给予B族维生素、辅酶Q10等治疗,1 y后随访患者情况较前无明显变化。
2 讨 论
1998年Hu等[2]发现ALS中约10%的患者存在特殊的临床表现,主要以对称性双上肢近端为主的肌无力和肌萎缩为主要特点,其他区域无或轻度受累,病情缓慢进展,临床症状体征局限在双上肢的时间大于12 m[3],患者呈现肩部下沉,双上肢旋前的特征性姿势,命名为FAS,被认为是ALS的变异型。FAS病因和发病机制尚不明确,有研究提示可能与超氧化物歧化酶-1基因突变有关[4],而核不均一核糖核蛋白A1(heterogeneous nuclear ribonucleoprotein A1,hnRNPA1)基因突变可能与家族FAS有关[5]。另有报道FAS合并干燥综合征及HIV的病例,提示其发病可能与免疫、感染因素等有关[6,7]。
FAS发病年龄多在50岁~60岁,男女发病比例约为(4~10):1[8],FAS患者三角肌、冈上肌、冈下肌、胸锁乳突肌和小圆肌发生明显萎缩。有研究显示FAS虽最终发展为以对称性双上肢近端为主的肌无力及萎缩,但有76%的患者首发症状为上肢远端或近端及远端均无力,并且双侧不对称受累,24%的FAS患者以近端肌无力起病,且只有3%的患者为对称性双上肢近端发病。其首发症状多样,可能是此病早期易误诊的原因之一。研究发现,FAS首发症状多发生在右上肢,即多数人的优势侧肢体,可能是由于优势侧肢体出现异常时更容易被发现所致[1]。本文中3例患者均为男性,除病例3患者发病年龄高于常见发病年龄,其他2例患者发病年龄基本与此病吻合。3例患者均有不同程度近端肌萎缩,其中2位例患者以右上肢起病,1例患者以双上肢起病。另外,本文3例患者病程分别为3 y、14 m、15 m,均无其他部位肌肉损伤的症状体征,符合FAS的特征。
FAS患者多数表现为上肢的下运动神经元损伤,研究发现70%的FAS患者下肢逐渐出现上运动神经元受累表现,而只有50%的患者上肢有上运动神经元损伤的体征[8]。可能是由于双侧上运动神经元损伤的患者由于损伤对称,不易被察觉,多在疾病进展时就诊,此时锥体束征常常被肌萎缩掩盖。而以单侧上运动神经元损伤起病的患者,多因损伤不对称而较早察觉,此时体格检查可以发现上运动神经元损害表现[9],可见上肢的病理反射或深反射活跃,但不会出现上肢的肌张力增高或者肌阵挛[10]。从表1中可见3例患者上肢均有下运动神经元损伤体征,1例患者左上肢Hoffmann征阳性,3例患者双下肢均无损伤体征,符合FAS常见体征。
FAS的进展缓慢,损伤区域局限于颈区的时间较长,肌电图常表现为上肢近端肌肉的神经源性损害,继发的轴索损害也以上肢为主,表明其病变范围相对局限[11]。诊断需满足肌电图中延髓、颈、胸、腰骶4个区域中3个区域出现失神经电位,研究显示约27%~77%的FAS患者逐渐出现下肢及球部受损的肌电图表现[3]。本文3例患者中1例肌电图存在3个区域失神经电位,2例肌电图存在2个区域失神经电位,考虑与病程较短有关。FAS骨骼肌病理改变特点为小角化肌纤维、“靶纤维”、肌原纤维网紊乱等神经源性损害表现。血肌酸激酶可轻中度升高或正常,考虑与肌肉无力导致活动后过劳有关[12]。有报道称FAS患者的颈椎MRI中T2加权相可出现高信号病灶,在轴位上呈“蛇眼征”,但此征象并不特异,部分平山病、进行性脊肌萎缩患者的颈椎MRI也可有此改变[13],考虑是脊髓前角细胞的萎缩及神经胶质细胞增生所致。本文中3例患者1例行肌肉活检显示神经源性损害,2例未行肌肉活检。2例患者肌酸激酶升高,1例正常。3例患者颈椎MRI髓内未见异常。结合上表中3例患者各临床特点,考虑符合FAS诊断。
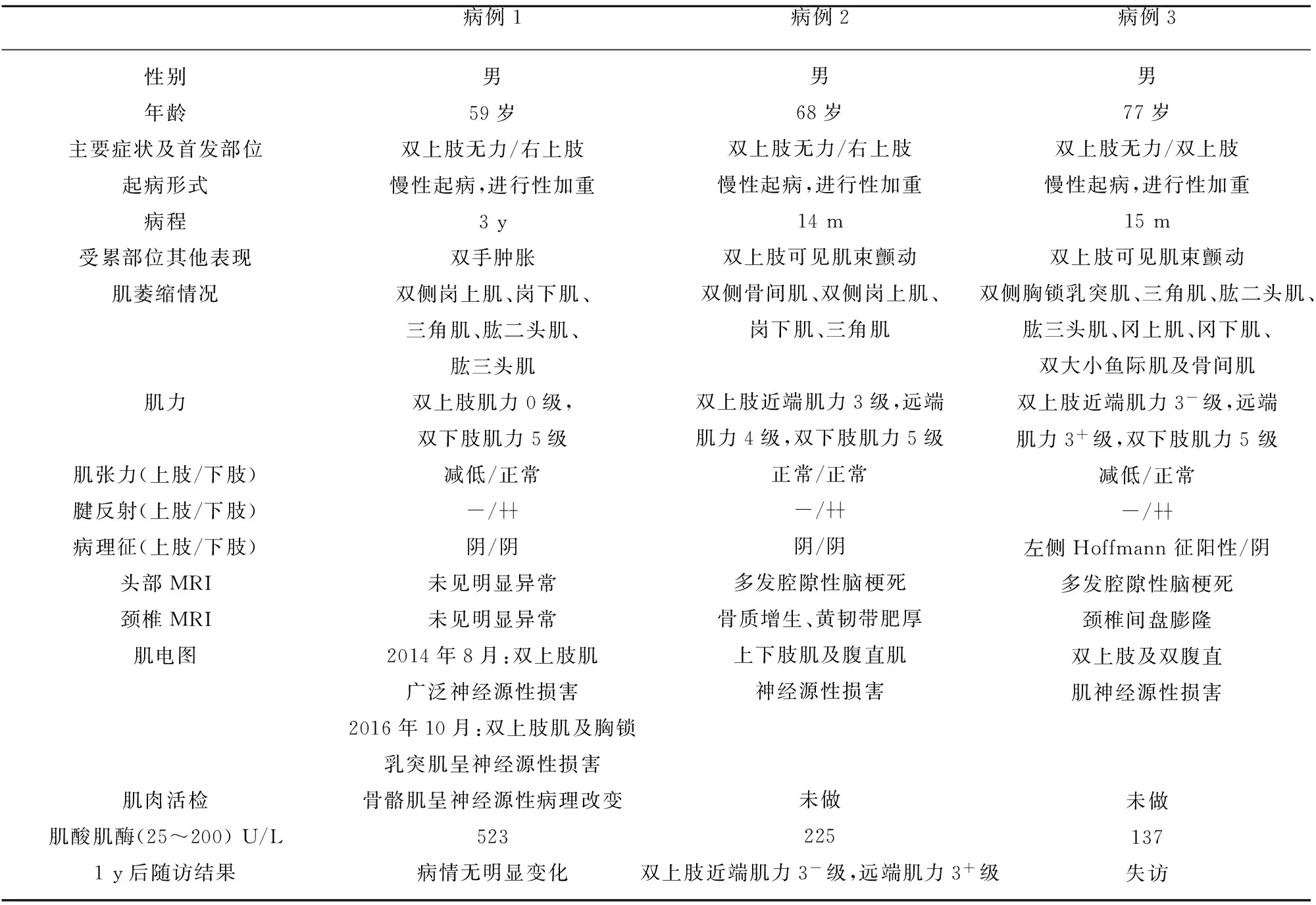
表1 3例FAS患者临床资料
诊断FAS时需排除平山病、脊髓型颈椎病、腕管综合征及多灶性运动神经病等疾病。平山病多发生在青年男性,不对称的上肢远端起病,颈椎MRI可见颈部生理曲度消失、非对称性萎缩,而屈颈位可见硬膜囊后壁前移、颈髓下段硬膜外强化信号及硬膜外后侧血管流空信号。脊髓型颈椎病及腕管综合征可分别通过颈椎MRI及肌电图鉴别。多灶性运动神经病被认为是自身免疫相关的周围神经病,多为非对称肢体无力,血清抗GM1特异性IgM抗体多升高,肌电图可见常见受压部位之外发生的运动传导阻滞,且免疫球蛋白治疗有效。上述疾病均为可治性疾病,因此诊断FAS时需仔细鉴别。
另外,FAS仍需与进行性脊肌萎缩及ALS相鉴别,三者均属于运动神经元病,但预后却不同,FAS预后相对较好[3]。FAS局限在双上肢的症状大于12 m是与其他两种疾病的一个重要鉴别点[3],除此之外,ALS的上、下运动神经元损伤体征均较明显,进展较FAS快,呼吸障碍出现较早,生存期较FAS短,有研究称ALS从发病到需要辅助通气约22 m,而FAS约为51 m[3]。有报道称ALS患者可见分裂手现象,即拇短展肌和第一骨间背侧肌更早出现肌萎缩无力且受累程度更重,而小指展肌相对保留,但FAS患者无此现象[8,14,15]。近年来,研究发现血清中神经丝蛋白浓度在ALS患者及健康人中有差异,其敏感性和特异性高达90%,脑脊液中的神经丝蛋白浓度可区分ALS及与其症状相似的其他疾病,敏感性达77%,特异性达88%,其浓度可能与疾病严重程度相关[16~19]。目前有关生物学标志物的研究仍在进行,如能够发现ALS及FAS的生物学标志物,则可使诊断更为便捷,在一定程度上可减少误诊及漏诊。
本文3例患者在营养神经及对症治疗后症状未见好转,1例患者病情较前进展。目前仍没有特别有效的药物阻止FAS病情进展,利鲁唑经临床证实能改善ALS患者生存期和推迟呼吸衰竭发生,但应用于FAS的效果仍有待观察。熟悉此病并早期诊断对明确患者预后有重要意义。
[1]Hubers A,Hildebrandt V,Ludolph AC,et al. Clinical features and differential diagnosis of flail arm syndrome[J]. J Neurol,2016,263(2):390-395.
[2]Hu MT,Ellis CM,Al-Chalabi A,et al. Flail arm syndrome:a distinctive variant of amyotrophic lateral sclerosis[J]. Journal of Neurology,Neurosurgery,and Psychiatry,1998,65(6):950-951.
[3]Wijesekera LC,Mathers S,Shaw CE,et al. Natural history and clinical features of the flail arm and flail leg ALS variants[J]. Neurology,2009,72(12):1087-1094.
[4]Valentino P,Conforti FL,Clodomiro A,et al. Brachial amyotrophic diplegia associated with a novel SOD1 mutation (L106P)[J]. Neurology,2005,64(8):1477-1478.
[5]Liu Q,Shu S,Peng B,et al. Whole-exome sequencing identifies a missense mutation in hnRNPA1 in a family with flail arm ALS[J]. Neurology,2016,87(17):1763-1769.
[6]Takakura Y,Murai H,Furuya H,et al. A case of brachial amyotrophic diplegia accompanied with Sjogren’s syndrome presenting good response to immunotherapies in the early course of the disease[J]. Clinic Neurol,2005,45(5):346-350.
[7]Berger JR,Espinosa PS,Kissel J. Brachial amyotrophic diplegia in a patient with human immunodeficiency virus infection:widening the spectrum of motor neuron diseases occurring with the human immunodeficiency virus[J]. Arch Neurol,2005,62(5):817-823.
[8]Yang H,Liu M,Li X et al. Neurophysiological differences between flail arm syndrome and amyotrophic lateral sclerosis[J]. PloS One,2015,10(6):e0127601.
[9]徐迎胜,张 朔,陈君逸,等. 连枷臂综合征颈区上运动神经元损害的检测[J]. 中华神经科杂志,2017,50(2):116-119.
[10]王秀丽,叶静纳,董 铭,等. 连枷臂综合征1例报告[J]. 中风与神经疾病杂志,2015,(10):948.
[11]徐迎胜,樊东升. 连枷臂综合征的临床和神经电生理研究[J]. 中华医学杂志,2013,93(1):23-25.
[12]沈宏锐,靳陶然,胡 静,等. 连枷臂综合征临床与病理分析[J]. 国际神经病学神经外科学杂,2014,(1):20-22.
[13]Sasaki S. Sporadic lower motor neuron disease with a snake eyes appearance on the cervical anterior horns by MRI[J]. Clinic Neurology Neurosurg,2015,136:122-131.
[14]方 佳,刘明生,管宇宙,等. 肌萎缩侧索硬化患者分裂手现象分析[J]. 中华神经科杂志,2016,49(3):227-231.
[15]Sun X,Zhang Z,Liu N. Absence of split hand in the flail arm variant of ALS[J]. Clinic Neurophysiol,2016,46(2):149-152.
[16]Roche JC,Rojas-Garcia R,Shaw CE,et al. A proposed staging system for amyotrophic lateral sclerosis[J]. Brain,2012,135:847-852.
[17]Steinacker P,Feneberg E,Kubisch C,et al. Neurofilaments in the diagnosis of motoneuron diseases:a prospective study on 455 patients[J]. J Neurol Neurosurg Psychiatry,2016,87(1):12-20.
[18]Puentes F,Topping J,Malaspina A. Immune reactivity to neurofilament proteins in the clinical staging of amyotrophic lateral sclerosis[J]. J Neurol Neurosurg Psychiatry,2014,85(3):274-278.
[19]Gaiottino J,Norgren N,Lindberg RL,et al. Increased neurofilament light chain blood levels in neurodegenerative neurological diseases[J]. PloS One,2013,8(9):e75091.
《医学参考报神经内科频道》征订启事

《医学参考报神经内科频道》微信二维码
《医学参考报神经内科频道》于2010年5月创刊。该报是由国家新闻出版总署批准、中华人民共和国卫生部主管、吉林大学白求恩第一医院神经内科承办的国际医学信息参考类报纸,报号CN 11-0269。
本报是国内首家神经内科专业性报纸,编委团队汇集国内顶尖神经病学专家,代表了国内神经内科领域的最高水平。本报致力于促进、活跃学术交流,快速准确的报道全球神经病学领域中的新理论、
新技术、新经验、新成果、新进展,推动我国神经病学的发展和有关专业队伍素质的提高,帮助临床医师选择“最适合”的专业信息,为他们的科研和临床提供重要的决策依据,更好地为广大人民群众的健康服务。
我们将以科学的精神和实事求是的态度办好医学参考报神经内科频道,倡导学术民主,鼓励学术争鸣,搭建多学科协作交流的平台,我们希望广大同仁能和我们一起通过本报来传递神经病学的最新信息,通过本报来了解神经病学的发展前沿,为我国神经病学事业贡献力量。
本报为双月刊,全年6期,8开8版,每期定价5元,全年30元。欢迎从事神经病学的医学工作者和相关人员,神经内外科、内科、儿科的临床医师、研究生、进修生及基础医学的教师、研究生订阅本报。
汇款地址:吉林省长春市东民主大街519号,《中风与神经疾病杂志》编辑部,邮编:130061。
联系电话:0431-88782862、88782863。
猜你喜欢
保健与生活(2022年13期)2022-07-06
体育科技文献通报(2021年12期)2021-12-18
疯狂英语·新阅版(2021年8期)2021-09-10
考试与评价·高二版(2020年2期)2020-09-10
科学之谜(2019年3期)2019-03-28
北京广播电视报(2019年8期)2019-03-27
武警医学(2019年2期)2019-03-05
体育科技文献通报(2017年4期)2017-11-27
上海医药(2017年2期)2017-03-01
中国实用医药(2016年26期)2016-11-07
