
新时代下药品监管模式的探讨与实践
2018-03-11 19:30曹萌王冲付秋雁谭建新张一琼丁立承于玲莉刘凌毅吴浩李建平
上海医药 2018年3期
关键词:生命周期
曹萌+王冲+付秋雁+谭建新+张一琼+丁立承+于玲莉+刘凌毅+吴浩+李建平
摘 要 随着我国药品审评审批制度改革的不断推进,药品监管科学与事业的发展进入了新时代。本文结合改革创新的时代背景,概述我国目前的药品监管模式及其存在的缺陷,分析当前药品监管的形势,对药品监管模式进行探讨,并对欧美国家的药品监管情况作了简要介绍。本文还介绍了生物药新的监管模式的初步实践情况,对药品监管模式的创新探索作了展望。
关键词 药品监管模式 生命周期 生物药
中图分类号:R951 文献标识码:C 文章编号:1006-1533(2018)03-0005-05
Discussion and practice of drug regulation mode in new era
CAO Meng, WANG Chong, FU Qiuyan, TAN Jianxin, ZHANG Yiqiong, DING Licheng, YU Lingli, LIU Lingyi, WU Hao, LI Jianping*
(Shanghai Center for Drug Evaluation and Inspection, Shanghai 201203, China)
ABSTRACT Along with the comprehensive progress made in the reform of the drug review and approval system in China, the development of the science and undertakings of drug regulation has been entered into a new era. The current regulation mode in China and its imperfections are summarized by combining the era backgrounds of reform and innovation, the current situation of drug regulation in China is analyzed and the advantages and disadvantages of this drug regulation mode are discussed as well as the drug regulation status in European Union and the United States is briefly introduced. Meanwhile, the tentative practice of new regulation mode for biologics in China is introduced, and the innovative explorations of drug regulation mode is also prospected.
KEY WORDS drug regulation mode; life-cycle; biologics
藥品作为一种高风险商品,与人民群众的身体健康、生命安全关系密切,如何对其进行科学、有效的监管,长期以来一直受到社会各界的高度关注,各国政府对药品也都以慎重的态度进行严格的监管[1]。近年来,我国的药品质量和标准不断提高,药品研发和生产能力发展迅速,公众用药的需求得到了较好的满足,但药品注册、生产、流通和用药安全方面存在的问题也日益凸显[2]。党和国家非常重视药品的监管情况。2015年5月,中共中央总书记习近平在主持健全公共安全体系的中央第23次集体学习时强调,要切实加强食品和药品的安全监管,用最严谨的标准、最严格的监管、最严厉的处罚、最严肃的问责来加快建立科学、完善的食品和药品安全治理体系,严把从农田到餐桌、从实验室到医院的每一道防线。2015年8月,国务院办公厅印发了《国务院关于改革药品医疗器械审评审批制度的意见》,药品监管制度改革进入了新的快车道。2017年10月,中共中央办公厅、国务院办公厅联合印发了《关于深化审评审批制度改革、鼓励药品医疗器械创新的意见》,对在药品和医疗器械审评审批制度改革不断深化过程中备受关注的突出问题提出了明确的监管方向。
药品作为一种特殊商品,具有商品的生命周期属性。从研发立项到产品退市,对药品全生命周期的严格监管都是十分有必要的。从国内外的经验来看,实施以品种为主线、涵盖其全生命周期各个环节的安全信息和风险管控、将品种监管和体系监管相融合的监管模式具有一定的优势,是近年来药品监管模式的一种发展趋势。这种监管模式区别于分阶段的过程监管模式,将药品研发至销售过程中的各个过程监管条块有机结合,形成一个完整的链条,统筹安排、整体把控、持续动态管理。在新监管模式的实践中,逐步实践,完善制度,通过改革创新,进一步将监管范围从研发至销售拓展为研发—销售—不良反应监测—退市的全生命周期监管模式,这是新时代下药品监管模式的发展方向。
结合上海市生物药产业,特别是单克隆抗体类药品研发、生产公司和品种数以及技术水平均在全国处于比较领先的地位这一实际情况,上海药品审评核查中心在探索生物药新的监管模式方面进行了初步实践并取得了一定成效。以此为背景,本文对新时代下药品监管模式进行探讨,并介绍我们在生物药新的监管模式方面的初步实践情况。
1 目前的药品监管模式及其缺陷
我国目前的药品监管模式的特点是分阶段的过程监管。在药品生命周期的不同阶段,其监管过程大体上由国家和省级药品监管部门不同的处室及其对口技术支撑部门各自分别进行[1]。也就是说,监管过程实际上划分成了药品注册、药品生产、药品流通、不良反应监测等若干监管业务条块。
在分阶段的过程监管模式下,各个监管部门负责各自业务条块的监管工作,不同监管部门之间缺乏有效的沟通,各监管部门与监管和服务对象之间亦缺乏有效的沟通,由此可能造成信息和知识闭塞,各业务条块的监管也容易产生监管漏洞,达不到理想的监管效果,从而影响相关法律法规的严格落实,现有监管资源的作用也难以得到充分发挥[3]。此外,除GMP认证、跟踪和专项检查以外,我国目前对药品的监管还包括研制现场核查、临床数据核查以及各种专项检查等,这些核(检)查大多尚未明确以GMP为依据,药品生产与研发注册的监管尺度不统一,存在一系列的潜在风险。另外,我国参与药品监管的人员来自中央和地方各级药品监管部门,专业能力参差不齐;检查人员中有很多是兼职工作,经验不足,出勤和保障方面也存在问题;资料的审评、检查结果的量裁、整改情况的审核均可能存在尺度和水平不统一的情况,难以全面保证药品监管的质量;对检查员的培训、考核和奖惩制度亦较难整体统筹,不利于建立一支稳定的职业化、专业化检查队伍,影响了我国药品监管向世界先进水平靠拢的步伐。endprint
由此可见,我国目前的药品监管模式在推进药品全生命周期监管的规范化和医药行业整体发展方面存在一定的局限性。
2 新时代下药品监管的形势
医药行业被称为“永远的朝阳行业”。自改革开放以来,随着国内人民生活水平的持续提高和医疗健康需求的不断增加,我国药品需求增长迅速。在国内市场需求释放的刺激下,在不断增加的境内外投资带动和政府各项政策的扶持下,我国医药工业的经济增长率已连续多年远高于整体经济的平均增长率。尤其是近些年,我国医保体系不断健全,居民支付能力提高,对高质量医疗健康服务的需求明显增加,公众用药需求已从有药可用、用得起药向用放心药、优质药、创新药转变。2017年城市公立医院逐步取消药品加成,医疗体制改革取得实质性进展,以药补医的机制正被破除,公立医院运行朝着维护公益性、调动积极性、保障可持续性的方向发展。进入新时代后,以往药品创新不重视、仿制药质量良莠不齐、抗生素和一些注射剂使用过度等情况明显改变,以单克隆抗体类药品为代表的高新技术生物药、以嵌合抗原受体T细胞治疗为代表的先进个体化医疗产品、质量和疗效与原研药一致的高质量仿制药正在逐步进入临床应用。药品的关键质量属性、关键工艺参数、特有理化性质和药理毒理特征数据与其销售、使用、不良反应等方面的风险管控密不可分。医药行业发展的新形势对我国现在的分阶段过程监管模式提出了挑战,也凸显了向新的监管模式转变并进而完善药品全生命周期监管的必要性。
当前,我国药品监管机构实行国家和省级及以下垂直管理,地方药品监管部门同时接受上级药品监管部门和地方政府的双重领导。由于地区发展的不均衡,各地药品监管资源和效率存在一定的差异[4]。我国药品监管以中央审批、地方监督的模式为主,中央到地方的各个业务条块虽能处理各自监管环节内的问题,但往往难以按照品种特点构建起强大的监管链条,在事前预防、事中监督、事后惩戒和基于品种进行事件总结方面的效果有待提高。此外,监管经验也较难形成系统的知识库,较难有效促进监管水平和制度的创新。新时代下我国药品监管改革创新的形势是严峻的。
在我国医药行业高速发展、监管制度改革深入推进的新形势下,为保证药品质量和鼓励创新,国家食药监总局不断深化审评审批制度改革,从多方面发力、多点突破向纵深推进,出台了一系列新的举措。2015年以来国家食药监总局研究出台了一系列的规范性文件和征求意见稿,进一步将药品监管和行业发展推向全面规范和有法可依、有章可循的轨道。《药品管理法》和《药品注册管理办法》正在修订中,新的法律法规将从目前药品和企业捆绑、以生产企业为中心的监管状况转向秉持以上市许可持有人为责任主体、围绕药品进行监管的监管理念,凸显以品种为核心、统筹药品全生命周期的新的监管模式的发展方向。通过对新的监管模式的探索,逐步实现药品全生命周期监管,以适应新时代下患者用药需求和医药行业发展。
3 新的药品监管模式的探讨
药品从研发、生产、销售、上市后评价和不良反应监测到退市,其全生命周期是一个有机整体。条块化监管虽细分了药品生命周期的各个过程,但也阻碍了各个监管职责阶段之间的信息共享,无法很好地实现全过程监管。如果监管能把药品全生命周期中的研发、生产、流通、使用、不良反应监测和退市各个阶段的节点整合在一个链条上,通过融合监管部门内部的各职能条块,收集贯穿于研发、生产、流通和使用各环节的药品信息,包括同类品种特有的风险相关信息,就可更准确、更全面地把握药品风险,对药品形成更有效的监管。
对新的药品监管模式的探索,我们主要关注了以下几个方面:
一是以品种为核心,药品生命周期各阶段监管有机融合。药品从研发到退市的全生命周期是一个有机整体,监管的信息、人员、思路等不应受限于业务条块。药品知识和监管信息应全链条共享,使企业和监管机构在药品生命周期的所有阶段都能作出快速、知情的决策。
二是监管工作规范化、体系化。按照质量管理体系建设原则,在现有法律法规和制度体系文件的基础上,探索增设新的监管模式的配套部门制度,形成整套工作文件,并在执行过程中不断加以丰富和改进。建立试点工作组,设立工作目标,明确岗位职责,建立工作程序、作业指导书和统一的记录表单,不再按业务条块拆分业务部门及人员,不再按药品生命周期阶段各自建立监管工作要求,而是形成一系列适合药品生命周期全过程监管且可复制、可推广的配套制度、程序和记录。
三是建立药品生命周期档案(“一品一档”),全面整合品种数据、监管信息和知识库。以具体品种为主线,贯穿研发、临床、注册、生产、流通、不良反应监测和退市各个阶段的安全信息和风险管控,将品种监管和体系监管相融合,实现药品“一品一档”的构建和管理,并通过品种档案形成药品监管的大数据平台,为新的监管模式提供技术储备和信息支持,并使之成为药品监管知识库的一个重要组分[5]。
四是培养一专多能的复合型全职检查队伍,建立与药品全生命周期监管相适应的监管力量。明确试点工作组的框架及崗位职责,整体上符合相关专业背景要求,且在某一领域有较深厚的专业基础,是该领域高水平的专家,同时也具备药品研发、生产、流通和上市后评价全链条各环节监管工作的能力。监管人员应既是法律法规方面的专家,也是核查检查方面的专家,同时还应是特定学科专业领域的专家。
五是建立与监管和服务对象沟通的机制,规范与服务并举,促进信息交互,提高监管效能。以自愿申请为原则,构建透明、高效、专业的沟通机制,对药品生命周期各阶段的具体问题,由试点工作组统一对接、统一标准、统一流程、统一讨论和答复。试点工作组“一个窗口对外,一站式服务”,不再区分研发和注册、生产、流通、不良反应监测这些业务条块,充分体现新的监管模式的特点。
4 欧美国家药品监管模式简介
我国药品监管正在积极地与国际先进水平接轨。2017年6月,国家食药监总局宣布我国已正式加入人用药品注册技术要求国际协调会,这是一个具有里程碑意义的事件,也是我国医药产业向国际化方向发展的一个契机。通过借鉴国外相关的法规和指南,对探索我国药品新的监管模式具有重要的意义。endprint
欧洲药品管理局(European Medicines Agency, EMA)是欧盟的一个实体机构,1995年开始运行。欧盟的药品审评方式分为集中审评程序、分散程序、互认程序和单一成员国审评程序[1]。按照集中审评程序申报的药品可在通过上市许可后投放整个欧盟市场;分散程序则以一国作为申报审评的相关成员国,其他成员国审核、承认参照国的决定,或由申报方要求EMA仲裁;互认程序适用于大多数常规仿制药品,申报方可将一成员国的上市许可用于申报其他成员国认可;单一成员国审评程序许可的药品只能在单一成员国上市,仅需向单一成员国申报审评,手续相对简单,但此类申报情况相对较少。几乎所有的生物药及其他先进的治疗型医药产品均需按照集中审评程序申报。
欧盟的药品监管检查一般由EMA统一协调各成员国监管力量进行,相关的技术性问题解释以及法规制定也由EMA统筹。欧盟的检查员为专职人员。欧洲药品评价局检查处每年召开4次欧盟各国检查员代表碰头会,交流、沟通情况,研究工作中碰到的各种问题,对药品相关的法规和指南提出修订意见。这种会议也是一定形式的培训,对统一欧盟检查标准起到了十分重要的作用。GXP(GMP、GCP、GLP)检查是集中式的,检查机构全面负责药品的实验室、临床、生产的检查且检查标准相同,在欧盟范围内可互认。
欧洲制药工业协会联合会联合药品供应链建立了一套在欧洲范围内通行的药品电子监管系统,即欧洲药品验证系统。该系统对单件药品赋码,采用“配药点验证”模式,通过强制实行发药前监管码信息验证,以实现对药品的安全监管及流向追踪。这种“一品一码”的模式在欧洲各国已基本实现了全覆盖,其多年来运行的有效性也得到了较广泛的认可。
美国FDA是美国卫生与公众服务部下辖的11个业务部门之一。FDA的门药品评价与研究中心、生物制品评价与研究中心拥有较健全的“检查审评员”(inspectorreviewers)管理体系和人员队伍,统筹药品的临床试验、上市申请、检验、上市后不良反应报告和相关科学研究等问题的处理,也负责GLP和GCP合规性检查,同时与FDA的其他部门密切合作,共同负责确认药品生产符合GMP要求。由于法律法规体系相对健全,美国没有GMP认证检查,药品生产企业也不需持有相应的证书,GMP实施主要依靠企业的自觉、责任感和法律意识。从药品研发注册开始,申报人的样品试制/生产就需符合GMP要求。FDA也会以GMP为基本依据进行现场检查,自始至终以确保药品关键质量属性的管控为目的,确保各类监管检查标准的尺度统一,避免因业务条块、阶段过程分离造成的监管效能割裂,保证药品全生命周期监管要求的持续性。美国的药品现场检查与药品注册审评工作是相互联系、紧密结合的,体现了美国药品全生命周期监管的理念[6]。
FDA法规事务办公室统一主导所有的外场活动,下辖227家办公室和13家实验室,遍布整个美国,并与所有FDA监管部门以及联邦、州、地方、部落、属地或外国公共卫生监管同行联合开展工作。FDA的地方办公机构工作独立于美国各州政府,形成了职权集中的全美范围内的药品监管权力网络。FDA只接受联邦政府的领导,其执法和检查过程不受地方政府的干涉[7-8]。FDA的大区办公室主要负责联邦整体检查工作的协调,也承担法律法规制定等职责,但不具体参与现场检查任务,而由地区机构开展监管检查。FDA的这种独立于地方的监管机构逐级管理模式加强了药品生命周期各阶段各监管部门的监管协作,在全美范围内统筹监管、统一标准、提高效能、预防风险。
FDA在药品上市前对其研发进行专业指导,上市后对其进行严格的监管。FDA的监管体系在事前、事中和事后是一体化的,将审评、监管和惩戒作为一个链条,按品种进行统筹,以事前预防为中心,在监管过程中充分关注品种的特点。FDA的药品检查人员需经过全面的系统化培训,同时具备审评与检查的资质。检查组采取系统化的、基于风险管理的方法开展检查,发现违规问题后主要以“警告信”的形式予以通报和处理,由技术审评人员和安全监管人员共同分析相应的缺陷并联合开展调查。对品种生命周期的监管情况,FDA监管链条各环节的专业人员会共同进行总结,并不断完善风险预防机制。
欧美的医藥行业及其监管起步早,经验积累相对丰富,医药体系和医药监管模式在全球处于领先地位,其理念和实践经验值得我们探讨和研究。通过对先进的监管模式的借鉴,结合我国国情有针对性的探讨,有利于促进我国药品监管模式与国际先进水平接轨。
5 生物药新的监管模式的初步实践
上海市近年来申报的单克隆抗体(以下简称为“单抗”)类品种有70余个,涉及单抗类品种研发的机构有20余家,具备单抗类品种生产能力的企业有8家,产业高度集中,在国内处于领先地位。如何指导单抗类品种研发、规范注册申报并最终加速市场急需品种的上市,这些都是上海市药品监管机构亟待摸索解决的问题,而探索新的监管模式是顺应当下蓬勃发展的生物药产业以及众多患者临床用药迫切需求的。在生物药全生命周期监管模式的探索方面,上海药品审评核查中心(以下简称为“我中心”)提出了新时代下新的药品监管模式方案,并专门组建了生物药小组,以生物药为试点开展了初步实践。
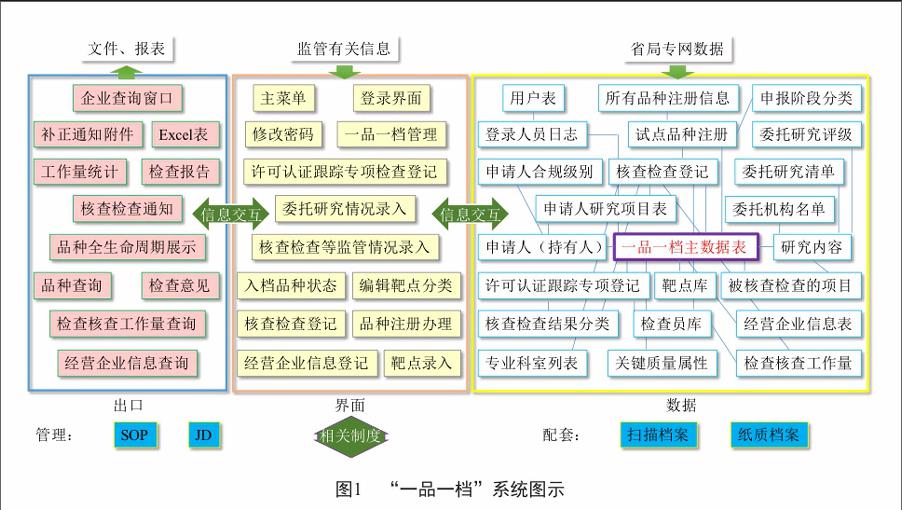
建立“一品一档”系统是实施新的药品监管模式探索的首要任务之一。我中心生物药小组首先明确了建设目标:覆盖各条块;衔接平稳、兼容过渡;可推广;便于改进;多重数据安全;表单关系清晰;窗口化、信息化;促进沟通、提高效能。随后,用2周的时间即实现了系统正式运行。“一品一档”系统将注册、生产、流通几大条块的过程监管有机融合成一个完整链条,并预留了不良反应监测和退市数据信息接入口,为扩展成为药品全生命周期监管系统做了准备。系统设置多级访问权限,具备较严谨的访问控制体系,以品种为核心,条理清晰。该系统包括近40个数据表,通过每个品种唯一的身份识别号对该品种的各项关键质量属性、监管信息和专业知识进行关联存储,同时设计了近50个功能窗口进行数据库管理,还有十余个标准化电子报表解决输出方案(图1)。“一品一档”系统规定品种档案包括关键的原料/辅料/包材质量标准、关键工艺的描述及其控制参数、品种关键的质量属性和其他与质量有关的关键内容,以及注意事项、药品说明等。在药品注册研发现场核查、临床研究核查、注册生产现场检查、药品生产企业检查、药品流通企业检查和药品不良反应报告中收集到的经过整理或消化的信息也及时录入“一品一档”系统,形成一套不断更新的监管品种知识库。endprint
上海某公司的某单抗注射液品种是我中心生物药小组试点新的药品监管模式的一个实例。该品种是以CD20为靶点研发申报的一种治疗用生物药,检查组在检查前调阅了“一品一档”系统知识库,以全面了解该品种的特点和风险把控重点。关键原辅料质量标准主要涉及细胞库、培养基、反应装置和处方等;关键工艺参数包括温度、搅拌、通气、营养、微环境、纯化、除病毒以及制剂和灌装等;关键质量属性主要为结构序列、纯度、组分分布和无菌等;作为生物药的共性特点,注意事项包括贮藏和运输等;以往对该品种研发、生产、流通全过程各环节的监管信息和重要风险还提示,涉及单抗类生物药研发方面的科学技术相关知识以及国内外同类型、同靶点品种的研发、申报、上市情况也需予以考虑。综合以上品种知识库信息,我中心生物药小组迅速组建了专项检查组,制定检查计划,确定检查要点,并明确了在检查中应同时关注历史核检查意见及整改措施。
顺利完成上述监管检查工作需要多方面的知识。为此,专项检查组在成员方面涵盖了注册、生产、流通、临床试验等多领域的共十余位检查员,提早成立、提前介入,并根据相应申报有计划地开展了生产许可检查、药学核查、临床试验核查和实验室核查。在申报人筹备申报期间,检查组统一接受了申报人对监管链条各环节问题的咨询,并尽可能发挥桥梁作用促进其充分、合理地准备与国家食药监总局的沟通,服务企业以提高申报工作效率。监管检查过程中,检查组派出多个小组赴多地并联进行药学、临床和样本分析现场核查。注册专业监管人员根据计划有针对性地对关键点的真实性、完整性和一致性进行核查;生产专业监管人员在检查过程中重点关注了设施、质量保证、人员和文件等体系方面的内容;流通专业监管人员结合药品稳定性试验数据、非临床和临床安全性评价数据,对药品上市后可能存在的运输、存储、处置和用药安全性等方面的风险提出意见。检查组整体统筹,汇总监管信息和检查结果,并及时纳入“一品一档”系统知识库。通过全面分析,生物药小组对该品种的整体情况出具了综合监管意见,这些意见可在科学性方面对国家食药监总局药品审评中心的技术审评起到协助作用,并为国家食药监总局整体统筹监管提供参考。
6 结语
药品审评审批制度改革进入了新的阶段。随着改革深入,更多的放心药、优质药、创新药有望成为公众可及的药品。为了进一步提高药品监管效能、规范行业整体协调发展、有效管控用药安全性方面的风险、推动我国药品监管与国际先进水平接轨并更好地契合药品上市许可持有人制度的全面实施,探索新的药品监管模式是很有必要、也是切实可行的。我们针对生物药新的监管模式的探索已取得初步成效,今后我们力争在进一步开展新的药品监管模式实践的基础上不断总结经验、改进流程,进行适用于更大范围、更多药品类别的可推广的药品监管模式的创新探索。
参考文献
[1] 王麟达, 陈伟, 陈桂良. 新药研发与法规管理[M]//白东鲁,陈凱先. 高等药物化学. 北京: 化学工业出版社, 2011: 504-532.
[2] 唐民皓. 药品安全:从“监管”向“治理”的转型[J]. 财经法学, 2015, 1(4): 40-48.
[3] 孙咸泽. 用现代信息科技磨砺监管利器——食品药品监管信息化的建设与发展[J]. 中国食品药品监管, 2012(3): 12-15.
[4] 唐民皓, 许瑾, 史岚, 等. 地方食品药品监管行政指导制度的构建与对策[J]. 中国卫生资源, 2011, 14(6): 379-383.
[5] Garg L, Elshorbagy S, Gupta V, et al. The impact of information systems on management performance in the pharmaceutical industry [J]. J Cases Inf Technol, 2015, 17(3): 56-73.
[6] 韩胜昔, 叶露. 中美两国药品安全监管体制的比较研究[J]. 中国药房, 2015, 26(10): 1309-1312.
[7] 高敏洁. 美国FDA对新药临床试验申办者和合同研究组织的监管模式[J]. 中国新药与临床杂志, 2016, 35(2): 109-113.
[8] 杨依晗, 王广平, 高惠君. 美国食品药品监管大数据实践简介及对我国的启示[J]. 上海医药, 2017, 38(9): 60-65.endprint
猜你喜欢
现代仪器与医疗(2022年2期)2022-08-11
中国外汇(2019年13期)2019-10-10
民用飞机设计与研究(2019年2期)2019-08-05
中国特种设备安全(2018年11期)2019-01-08
消费导刊(2018年10期)2018-08-20
领导决策信息(2017年16期)2017-06-21
中国工程咨询(2016年3期)2016-02-13
塑料包装(2015年2期)2015-12-20
地理与地理信息科学(2015年4期)2015-10-13
塑料包装(2015年1期)2015-09-26
