
反相高效液相色谱法测定治糜康栓中苦参碱和氧化苦参碱的含量
2018-03-02 09:18宋凤媛董坤园许天阳
长春中医药大学学报 2018年1期
于 澎,宋凤媛,钱 圳,董坤园,许天阳
(长春中医药大学药学院,长春 130117)
治糜康栓由黄柏、苦参、枯矾、儿茶、冰片5味中药组成[1],其中苦参为豆科植物苦参(Sophora fl avescens Ait.)的干燥根,主要活性成分为苦参碱及氧化苦参碱。
治糜康栓原质量标准中仅对黄柏中盐酸小檗碱进行了含量测定,但未说明苦参中活性成分的含量测定方法[2-3]。本文建立治糜康栓中苦参碱及氧化苦参碱总含量的测定方法,可以提升该制剂原有质量标准。
1 仪器与试药
LC-2030高效液相色谱仪(日本岛津公司)、十万分之一天平(METTLER TOLEDO);色谱柱4.6 mm×250 mm,ZORBAX SB-C18,5 μm(Agilent Technoligies);治糜康栓为市售品及自制品,苦参碱对照品(110805-200508)、氧化苦参碱对照品(110780-201508)由中国食品药品检定研究院提供。
2 方法与结果
2.1 治糜康栓的制备 1)市售栓剂:购自通化金马药业,批号20170530,保存备用。2)实验室自制治糜康栓:改进药典工艺路线,去掉醇沉步骤,其余步骤相同。2.2 苦参碱、氧化苦参碱的含量测定
2.2.1 色谱条件与系统适应性试验 填充剂:十八烷基硅烷键合硅胶;流动相:乙腈-磷酸盐缓冲液(7 :93,每1.7 g磷酸二氢钾加500 mL纯水溶解,用磷酸调节pH = 3);检测波长:220 nm;流速:1 mL/min;进样量:10 μL;2种成分理论板数均不低于2 000。
2.2.2 对照品溶液的制备 取苦参碱对照品、氧化苦参碱对照品适量,加流动相分别制成约0.2 mg/mL的苦参碱溶液及约0.1 mg/mL的氧化苦参碱溶液。
2.2.3 供试品溶液的制备 取上述2种栓剂,剪碎,每个样品约取3 g,置具塞锥形瓶中,加0.5 mL浓氨水,润湿后加20 mL三氯甲烷,称重,超声(功率250 W,频率50 Hz)25 min,放至室温,用三氯甲烷补足减失的重量,取上清液并过滤。取5 mL续滤液,以中性氧化铝柱为固定相(100~200目,5 g,内径为1 cm),依次用三氯甲烷20 mL、三氯甲烷-甲醇(7 : 3)20 mL洗脱,收集洗脱液并蒸干。用流动相溶解残渣,定容至10 mL,摇匀,保存备用。
2.2.4 测定方法 取上述对照品以及供试品溶液各l0 μL,依次注入高效液相色谱仪,测定。
2.3 方法学考察
2.3.1 线性关系 1)苦参碱线性关系:配制浓度为20、50、100、150、200 μg/mL 的苦参碱对照品溶液,按2.2.1项下色谱条件测定,得到苦参碱线性回归方程:Y=5 456.8X+5 992.5,r2=0.999 9;结果表明在20~200 μg/mL范围内呈线性。2)氧化苦参碱线性关系:配制浓度为10、25、50、75、100 μg/mL的氧化苦参碱对照品溶液,按2.2.1项下色谱条件测定,得到氧化苦参碱线性回归方程:Y=5 429.7X-1 164.6,r2=0.999 8;结果表明在10~100 μg/mL范围内呈线性。
2.3.2 精密度试验 取2.2.2项下对照品溶液,分别重复测定6次,10 μL/次。得到苦参碱与氧化苦参碱含量的相对标准偏差分别为0.04%、0.15%,表明仪器精密度良好。
2.3.3 重复性试验 精密称取实验室自制栓剂6份,按2.2.3项下制备方法制备,按2.2.1项下色谱条件测定,得出6份供试品中苦参碱及氧化苦参碱的总含量的相对标准偏差为3.84%,表明方法重复性良好。
2.3.4 稳定性试验 精密称取实验室自制栓剂,按2.2.3项下制备方法制备,按2.2.1项下色谱条件,于0、2、4、8、12、24 h后分析测定,得出苦参碱及氧化苦参碱总含量的相对标准偏差为3.63%,表明在24 h内供试品溶液稳定。
2.3.5 加样回收率实验 精密称取已知含量的实验室自制栓剂6份,每份约3.0 g,分别精密加入对照品适量,按2.2.3项下制备方法制备,按2.2.1项下色谱条件测定,得出实验室自制栓剂中苦参碱和氧化苦参碱的回收率分别为98.75%、99.46%,相对标准偏差分别为2.61%、2.29%,表明方法准确度良好。
2.4 含量测定实验结果 取2.2.3项下供试品溶液,按
2.2.1 项下色谱条件测定,测定结果,见表1。
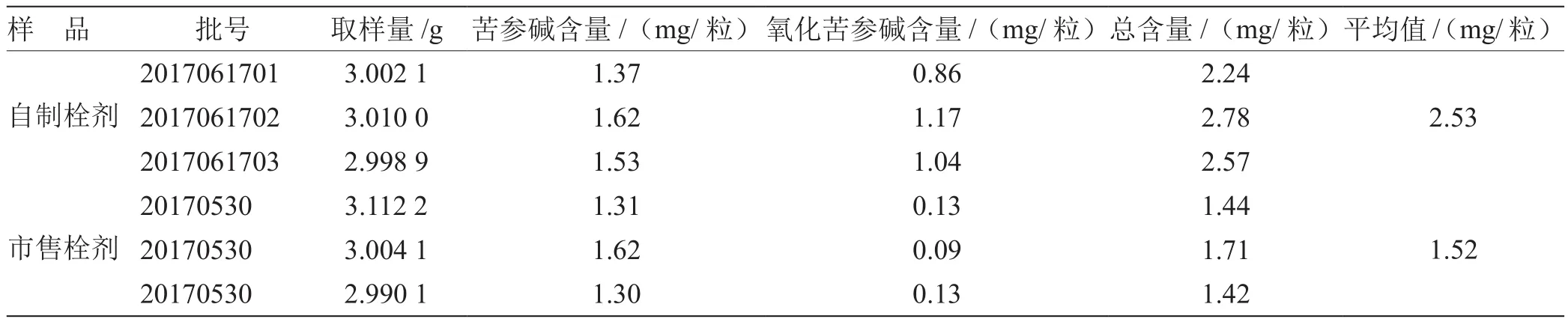
表1 实验室自制栓剂及市售栓剂含量测定结果
3 小结
3.1 色谱柱的选择 高效液相色谱法测定生物碱类成分,《中国药典》分别收载了十八烷基硅烷键合硅胶柱和氨基柱测定法,但氨基柱价格较高不常用。同时预试验结果表明其出峰时间过长,因此本试验选择了实验室常用的十八烷基硅烷键合硅胶柱,同时测定治糜康栓中苦参碱及氧化苦参碱的总含量[4]。
3.2 检测波长的选择 本试验先后以205、210、215、220 nm为检测波长,结果发现波长越小,生物碱的吸收峰越大,但杂质峰也随之明显,故本试验采用220 nm为测定波长。
3.3 供试品制备方法的选择 分别以二氯甲烷、三氯甲烷和乙醚作为提取溶剂,筛选后发现使用三氯甲烷并超声处理,提取效率最高[5-7]。浓缩方式分别采用蒸干、常温挥干2种,结果表明,干燥方法对含量无显著影响。为避免基线干扰,最终选择以流动相定容供试品。
3.4 流动相的选择 本次试验分别尝试了以甲醇-水(55 : 45)、甲醇-水-三乙胺(55 : 45 : 0.02)[8]、乙腈-水-三乙胺(20 : 80 : 0.02)、乙腈-磷酸盐(10 : 90)作为流动相;经研究发现,流动相为乙腈-磷酸盐缓冲液(7 : 93,1.7 g磷酸二氢钾加500 mL纯水溶解,用磷酸调节pH = 3)时,峰形较好,平衡时间最短,可以排除来自其他成分的干扰,目标组分可与样品中其他组分较好的分离[9-11]。
3.5 栓剂前处理工艺的优化 实验证明醇沉可能造成中药有效成分的流失,影响中药制剂的有效性,并且存在醇沉时间长等问题。本实验证明未经醇沉的栓剂中苦参碱和氧化苦参碱的总含量显著增多,可见除非实践证明醇沉法不影响有效成分含量和疗效,否则一般不宜采取醇沉法,以传承中医药传统特色[12]。
结论:本实验结果表明,单独将苦参碱或氧化苦参碱含量作为含量测定指标是不完整的,以它们的总量为定量指标更恰当[13-20]。本实验建立的方法可以同时测定治糜康栓中苦参碱和氧化苦参碱的含量,方法简便,结果准确,可以更加全面地评价治糜康栓的质量状况。由于本实验研究的样品数量少,尚无法制定该制剂的最佳质量标准,今后将逐步积累数据,为制定治糜康栓质量标准提供依据。
[1]国家药典委员会.中华人民共和国药典:1部[M].北京:化学工业出版社, 2015:111.
[2]李晓燕,赵韶华,等.苦参碱含量测定中色谱条件的改进[J].中成药, 2004, 26(2):172-173.
[3]罗明,贺平,吴孟超,等.高效毛细管电泳法测定苦参碱和氧化苦参碱[J].中草药, 1999, 30:(4):26.
[4]刘莹,孟庆妍,翟宏宇. HPLC法同时测定复方苦参肠炎康片中苦参碱和氧化苦参碱的含量[J].长春中医药大学学报,2011, 27(5):842-843.
[5]唐坤,李标,夏晨燕,等.反相高效液相色谱测定清益康乳膏中苦参碱的含量[J].时珍国医国药, 2007, 18(8):1829-1830.
[6]毛丹,陈钶. HPLC法测定苦参软膏中苦参碱、槐定碱和氧化苦参碱[J].中成药, 2011, 33(9):1531-1534.
[7]张蕾,陈晓辉. RP-HPLC法测定苦参中苦参碱的含量[J].沈阳药科大学学报, 2005, 22(1):33-35.
[8]吴春红,唐卫文. HPLC法测定苦参药材中苦参碱的含量[J].药物分析杂志, 2008, 28(7):1147-1149.
[9]时晓亚,方宝霞.高效液相色谱法测定复方苦参子洗液中苦参碱与氧化苦参碱的含量[J].新乡医学院学报, 2011,3(28):288-290.
[10]杨文远,杨宁莲. HPLC法同时测定苦豆子中苦参碱与氧化苦参碱[J].宁夏大学学报, 1996, 17(4):13.
[11]吴迪,梁健. RP-HPLC法同时测定复方苦参注射液中氧化槐果碱、氧化苦参碱和苦参碱的含量[J].沈阳药科大学学报, 2006, 23(4):220-223.
[12]石猛,莫尚志.醇沉法存在的问题及解决办法[J].中药材,1999, 22(6):313-314.
[13]陈宏降,李祥,陈建伟,等.中药三白草地上部位的化学成分研究(Ⅰ)[J].南京中医药大学学报, 2009, 25(4):286.
[14]邓惠文,欧子强,李元文,等.中药蛇床子中黄酮类成分的研究[J].广州化工, 2009, 37(6):105-106,114.
[15]孙文基,贾敏鸽.苦参及其复方中苦参碱与氧化苦参碱的转化研究[J].药物分析杂志, 2003, 23(2):90-93.
[16]江维克,王丽. HPLC法同时测定复方石韦片和苦参药材中苦参碱和氧化苦参碱的含量[J].中成药, 2005,27(1):45-48.
[17]杨志欣,王海威,张文君,等.苦参总黄酮含量测定方法的优化[J].中成药, 2017, 39(5):946-951.
[18]计曼艳,曾琨,王华. HPLC-DAD法同时测定中药苦参中七种生物碱成分的含量[J].中国药师, 2017, 20(1):60-63.
[19]付起凤,曹琦,吕邵娃,等.正交法优化苦参中苦参生物碱的超声提取工艺[J].中医药信息, 2015, 32(1):11-13.
[20]王秀兰,洪宗超,吴松涛,等.高效液相色谱法测定复方苦参汤中苦参碱、氧化苦参碱的含量[J].药物分析与检验, 2017, 15(10):1426-1429.
猜你喜欢
食品安全导刊(2021年36期)2021-03-14
科学导报(2020年75期)2020-12-21
中华养生保健(2020年4期)2020-11-16
氯碱工业(2020年6期)2020-03-01
中国外汇(2019年10期)2019-08-27
中国外汇(2019年10期)2019-08-27
中国外汇(2019年22期)2019-05-21
中国外汇(2019年21期)2019-05-21
家庭医药·快乐养生(2017年5期)2017-05-18
劳动保护(2010年8期)2010-11-11
