
共晶技术提高黄芩素溶出度及生物利用度的研究
2018-02-15 02:37:12周生研张博文魏元锋张建军
中国药科大学学报 2018年6期
周生研,张博文,魏元锋,钱 帅,张建军,高 缘*
(中国药科大学 1中药学院;2药学院,南京211198)
药物共晶是指将一种药物与另一种非活性或活性药物通过非价键以固定化学计量比结合在同一晶格中形成的晶体[1]。近几年来已成为改善药物的理化性质[2]、提高稳定性[3]和生物利用度[4],改善药效,获得新的适应症或降低不良反应的创新性技术[5],同时还有可能获得知识产权的保护[6-7]。例如,诺华公司研发经FDA和EMA批准上市的治疗心力衰竭的共晶制剂沙库比曲缬沙坦片(商品名EntrestoTM)就是一个典型范例[5],该产品成功运用共晶技术同时提高了两种生物药剂学分类系统(BCS)Ⅱ类难溶性药物的溶解溶出度,使其易于吸收,更好地发挥临床疗效。
黄芩素(baicalein,BE,图1-A)是天然植物黄芩的主要成分,现代研究表明,BE具有抗肿瘤、抗艾滋病毒、抗菌、抗肥胖、抗氧化、抗病毒等多种药理作用[8-14],目前在临床上主要用于抗菌消炎和抗感染,加之其对血液细胞及肝细胞等正常细胞无毒性,BE具有较好的临床应用价值及前景。然而,BE属于BCS Ⅱ类药物[4],亲水性差、溶解度低、口服生物利用度差,大大限制了其临床应用。目前,已有利用共晶技术来改善其溶解度和口服生物利用度的研究,例如黄芩素-烟酰胺共晶[4,15]。咖啡因(caffeine,CA,图1-B)是中枢系统兴奋药物,常用于减少疲劳和治疗困倦,属于GRAS(Generally Recognized As Safe)的小分子配体[16],具有多个吸电子基团,在共晶制备上具有优势,有研究表明咖啡因与杨梅素制备成共晶,可以显著地提高杨梅素的特性溶出速率[17]。因此,本研究选择咖啡因作为共晶形成物,与BE制备成共晶,旨在通过共晶技术提高BE溶出度和生物利用度。
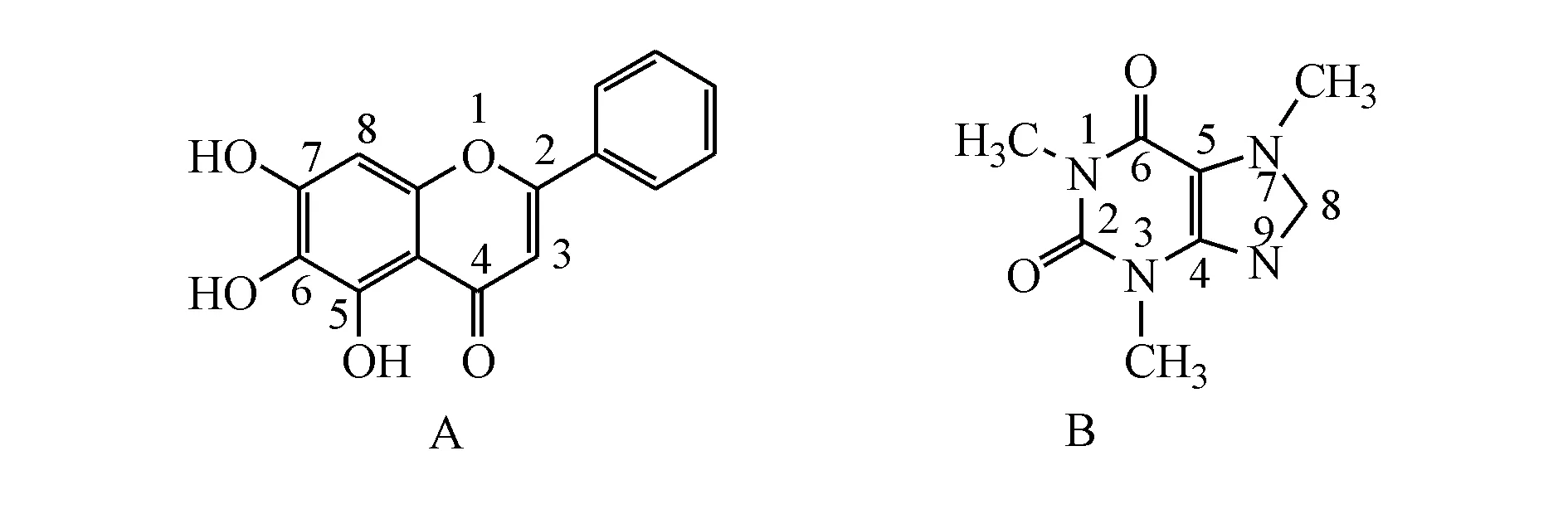
Figure1 Chemical structures of baicalein (BA,A) and caffeine (CA,B)
1 材 料
1.1 药品与试剂
黄芩素(BE,纯度98.58%)、黄芩苷(纯度98.97%)(南京泽朗医药科技有限公司);咖啡因(纯度≥99.5%,康丰生物科技);木犀草素(纯度99.3%,上海标准生物技术有限公司);羧甲基纤维素钠(上海国药集团化学试剂有限公司);抗坏血酸钠、肝素钠(185 IU/mg)(上海阿拉丁化学试剂有限公司);试验用水为采用Milli-Q水纯化系统(美国Millipore公司)制得;其他试剂均为市售分析纯。
1.2 仪 器
BS 110 S电子天平(德国Sartorius公司);Leica DM LM/P偏光显微镜(德国莱卡公司);差示扫描热分析仪(德国Netzsch公司);红外光谱仪(美国Thermo Fisher Scientific公司);D8 Advance X射线衍射仪(德国Burker AXS公司);高效液相色谱仪(SPD-10A检测器,LC-10AD泵,LC-Solution色谱工作站,日本岛津公司)。
1.3 动 物
实验用健康SD大鼠SPF级,均为雄性,体重230~270 g,南京青龙山实验动物中心提供,合格证号为SCXK(浙)2008-0033。
2 方 法
2.1 BE-CA共晶的制备
采用混悬液结晶法,将BE与CA按1∶1的物质的量比投料。称取黄芩素405.4 mg与咖啡因291.3 mg,置于一个合适大小的干净玻璃容器中。量取甲醇30 mL至容器内,用可透气的薄膜覆盖,于25 ℃条件下搅拌约5 h。将所得混悬液抽滤,所得产物于25 ℃条件下减压干燥12 h,即得。
2.2 BE-CA共晶的表征
2.2.1 热重分析(TGA) 采用热重分析法可以测定制备得到的共晶的晶体结构中是否含有溶剂分子。升温速率为20 ℃/min,范围为30~350 ℃。数据用热分析软件处理。
2.2.2 差示扫描量热法(DSC) 分别将BE、CA、二者的物理混合物(物质的量比1∶1)(PM)以及制备所得的共晶(BE-CA)样品过100目筛(约150 μm),备用。将这些样品分别置于铝坩埚中,升温速率为10 ℃/min,测定范围为25~300 ℃。数据用Netzsch-Proteus热分析软件(Version 4.2)处理。
2.2.3 傅里叶红外光谱法(FTIR) 分别将BE、CA、PM及BE-CA共晶与KBr压成薄片,在4 000~400 cm-1范围内进行扫描。数据用 Nicolet Omnic 红外光谱处理软件 (Version 8.0)处理。
2.2.4 X射线粉末衍射法(PXRD) 采用Cu-Kα靶,波长为1.540 562 Å,管压40 kV,管流40 mA,步长0.02°,扫描速度2°/min,扫描范围5~45°。
2.2.5 扫描电镜(SEM) 取BE-CA共晶样品适量,均匀散落于粘有黑色双面胶的载玻片上(厚度15~20 nm),激发电压控制20 kV,在真空条件下将试样表面喷金处理后,置于扫描电镜下观察,拍照,考察样品的外观和形态。
2.3 化学计量比的测定
称取BE-CA共晶10 mg于25 mL量瓶中,精密称定,加入甲醇溶解并定容至刻度,摇匀。精密移取上述溶液1 mL于10 mL量瓶中,加入流动相溶解并定容至刻度。采用HPLC法测定,以外标法测定并计算共晶中黄芩素与咖啡因的物质的量比。
HPLC系统采用Kromasil ODS C18色谱柱(4.6 mm×150 mm,5 μm),柱温为30 ℃;以甲醇-0.05%磷酸(70∶30)为流动相,流速1.0 mL/min;检测波长为275 nm,灵敏度0.05 aufs;进样体积 20 μL。黄芩素的线性范围是0.102~28.56 μg/mL,咖啡的线性范围是1.40~19.6 μg/mL。该方法分离度良好,重复性好、准确度高。
2.4 平衡溶解度的测定
分别测定BE和BE-CA共晶在水及各种pH缓冲液中的溶解度。分别量取水、0.1 mol/L磷酸盐缓冲液(pH 2.5、5.0、6.8、7.4)及醋酸盐缓冲液(pH 3.6、4.5)各5 mL置于西林瓶中,加入过量的药物后将西林瓶密封置于25 ℃恒温振荡器。振摇达到平衡后,取瓶中混悬液过0.22 μm水膜,滤液经适当稀释后进样进行HPLC分析,计算溶解度。
2.5 表观溶出速率
分别将BE、CA、PM和BE-CA共晶过100目筛,使样品粉末粒径范围均一。按照溶出度与释放度测定法《中华人民共和国药典》(2015年版)第二法(桨法)装置,转速50 r/min,在溶出杯中加入介质900 mL,于37 ℃平衡保温。溶出介质为0.01 mol/L HCl、水和磷酸盐缓冲液(pH 6.8)。精密称定相当于5 mg黄芩素的样品加入溶出介质中,于5、10、15、30、45和60 min取样,并及时补加等量的恒温介质,0.22 μm水膜过滤,采用HPLC测定溶出度。
2.6 特性溶出速率(IDR)
特性溶出速率的研究采用改良的Wood法进行[18],称取BE及BE-CA共晶约200 mg,用压片机压制成直径为8 mm致密规整的药片,其两个圆形表面的表面积为0.502 4 cm2。将药片的底面和侧面用蜂蜡模型包裹,使其只有一个圆形表面外露,可以与溶出介质接触。
溶出试验方法依照《中华人民共和国药典》关于溶出度测定的要求,在37 ℃条件下,将制备的用蜂蜡包裹的药片分别置于装有水、0.01 mol/L HCl和pH 6.8的磷酸盐缓冲液900 mL的溶出杯中,搅拌桨的转速为50 r/min。于5、10、15、30、45、60和120 min取样,并及时补加等量的恒温介质,0.22 μm水膜过滤,HPLC测定溶出度。用累积的单位表面积溶出量与时间绘制回归曲线,其斜率作为特性溶出速率,计算公式为:
IDR=(dW/dt)(1/S)=DCS/h
其中:IDR为特性溶出速率(mg·min-1·cm-2);dW为药物溶解的变化量(mg);dt为时间的变化量(min);S为药片的表面积(cm2);D为扩散系数(cm2·s-1);Cs为物质的饱和溶解度(mg·cm-3);h为扩散层的厚度(cm)。
2.7 BE-CA共晶的体内药代动力学研究
2.7.1 给药方案及样品采集 将SD大鼠随机分成3组,每组6只,实验前禁食12 h,可自由饮水,按剂量为每kg体重120 mg BE分别灌胃给予0.5% CMC-Na配制的黄芩素混悬液、物理混合物混悬液及黄芩素-咖啡因共晶混悬液,给药后分别于0,0.083,0.167,0.5,1,1.5,2,4,6,8,10,12,24 h从大鼠眼眶后静脉丛取血约200 μL,置肝素化离心管中,以10 000 r/min 离心5 min,取上层血浆,于-20 ℃冰箱保存。
2.7.2 血浆样品处理 取血浆100 μL,加入木犀草素内标溶液(152 μg/mL)5 μL和0.5 mol/L KH2PO4溶液50 μL(内含1%抗坏血酸钠),振荡混匀;加入乙腈150 μL,涡旋10 min,10 000 r/min 离心10 min,取上清液即得处理后的血浆样品。
2.7.3 HPLC色谱条件 采用Kromasil ODS C18色谱柱(4.6 mm×150 mm,5 μm),柱温为30 ℃;以甲醇-0.2%磷酸(57∶43)为流动相,流速1.0 mL/min;检测波长为276 nm,灵敏度0.05 AuFs;进样体积20 μL。色谱图中黄芩苷、木犀草素和黄芩素的保留时间分别为4.7、6.1和11.8 min,分离度良好且不被血浆内杂峰所干扰。黄芩苷及黄芩素分别在0.050 2~25.11 μg/mL(r2=0.998 6,n=6)及0.142 5~22.80 μg/mL(r2=0.996 4,n=6)范围内有良好的线性。BE的检测限及定量限分别为9.41和25.11 ng/mL,而BE的检测限及定量限分别为12.45和36.12 ng/mL。该方法回收率、精密度良好,样品稳定性高,可以满足测定需求。
2.7.4 数据分析 应用DAS2.0软件分析处理,采用统计矩法对灌胃给予BE,BE与咖啡因物理混合物(物质的量比1∶1)和BE-CA共晶的血药浓度-时间曲线进行分析。
3 结果与讨论
3.1 共晶的表征
3.1.1 热重分析法(TGA) HPLC测定结果表明,BE-CA共晶中的BE和CA百分含量分别为(58.36±0.80)%和(42.11±0.48)%,可推断在共晶中BE和CA的化学计量比为1∶1。
由于制备BE-CA共晶的溶剂为甲醇,属于无基因毒性的第二类溶剂。由TGA图2可知,共晶在200 ℃以上才开始失重,远高于甲醇的沸点(65 ℃),说明BE-CA共晶中不含甲醇溶剂分子,不是甲醇溶剂化物。
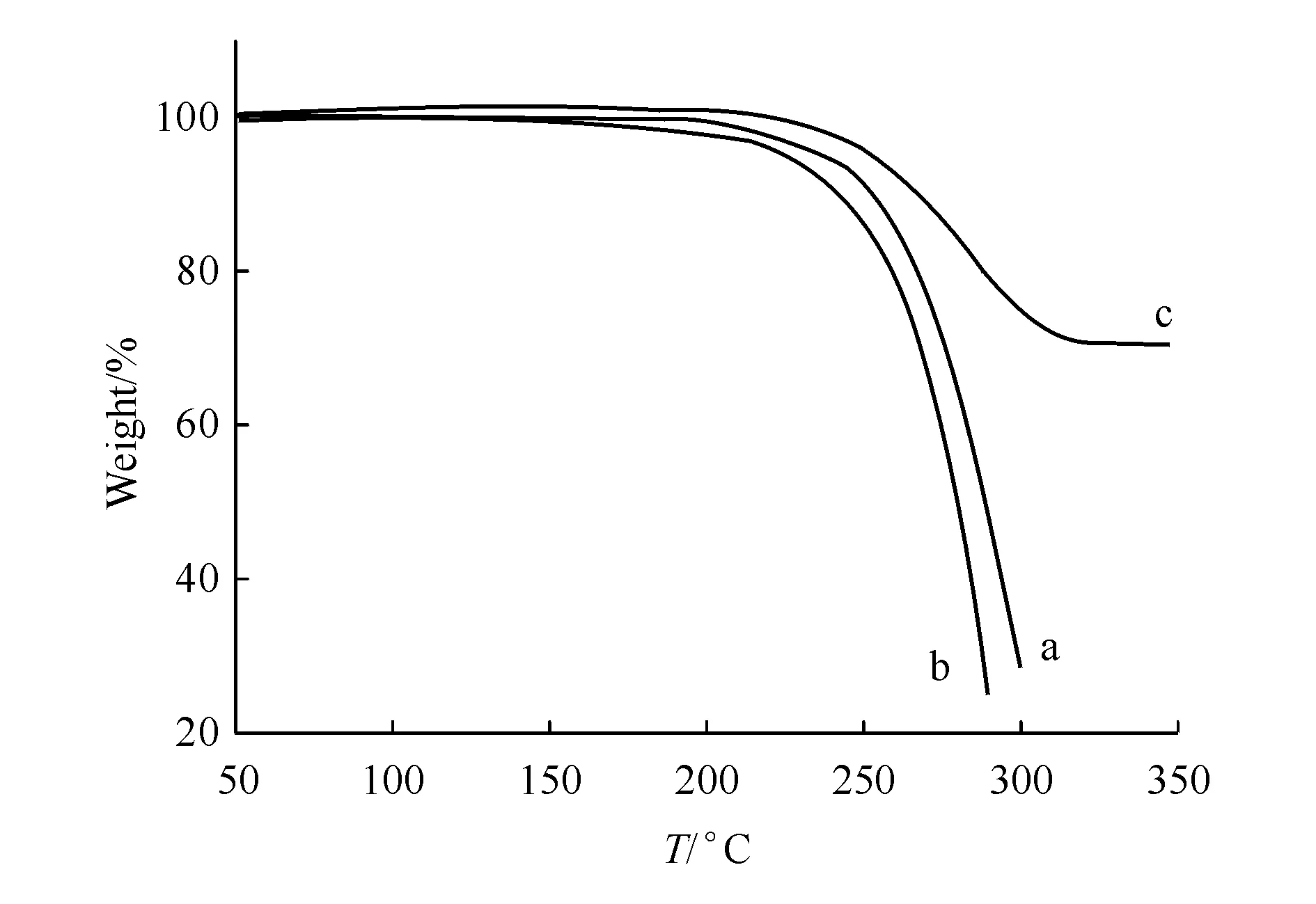
Figure2 Thermogravimetric thermogram of BE (a),CA (b) and BE-CA cocrystal (c)
3.1.2 差示扫描量热法(DSC)分析 由DSC图3可知,BE和CA分别在267.34 ℃和236.29 ℃处分别存在一个非常尖锐的吸热熔融峰,与文献报道一致[4,19]。物理混合物PM在180.03 ℃处有较宽的吸热熔融峰,可能是由于两者形成了低共熔物。而BE-CA共晶在199.27 ℃附近有单一且尖锐的吸热熔融峰,说明它是不同于两种单纯药物及其物理混合物的一种晶体新物质。
3.1.3 红外光谱(FT-IR)分析 由红外光谱图(图4)可知,BE的羰基峰υC=O位于1 656.9 cm-1,且酚羟基υOH在3 412.2 cm-1处峰形较宽,说明了BE本身的5-OH和4C=O 形成了分子内氢键[20];CA中羰基υC=O峰位于1 699.3 cm-1,υC=N峰位于1 657.8 cm-1,均与文献报道一致[21]。物理混合物PM的IR图为两种单组分谱图的简单叠加。

Figure3 Differential scanning calorimetry thermograms of BE,CA,physical mixture (PM) and BE-CA cocrystal
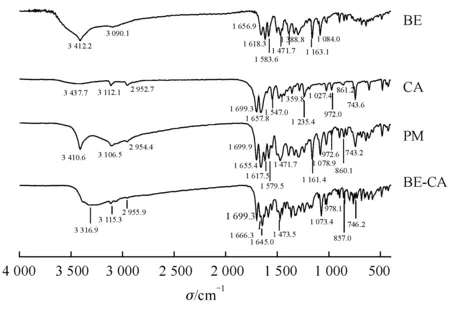
Figure4 FTIR spectra for BE,CA,PM and BE-CA cocrystal
由BE-CA共晶的红外光谱图可知,在形成共晶后,BE的酚羟基υOH峰由3 412.2 cm-1移至3 316.9 cm-1,且峰形变宽。同时,υC=O峰由1 656.9 cm-1红移至1 645.0 cm-1。说明BE中的酚羟基-OH、C=O的氧原子均可能参加了共晶中氢键的形成。CA中的υC=O峰一直保持在1 699.3 cm-1,未发生变化。但υC=N由1 657.8 cm-1移动到1 666.3 cm-1,8号位的γC-H由972.0 cm-1、861.2 cm-1移动至978.1 cm-1、857.0 cm-1,表明CA分子中C=N的N原子和=C-H的H原子都可能参与了氢键的形成,这与报道的CA-丙酸共晶形成相似[19]。
3.1.4 X射线粉末衍射法分析 由图5可知,BE的特征衍射峰在10.20°、11.32°、13.20°、15.32°、23.81°、26.18° 和 28.67° 2θ,CA的特征衍射峰在11.97°、23.99°、26.37°、26.98°和28.36° 2θ,均与文献报道一致[22-23]。物理混合物PM谱图为两种单组分谱图的叠加。而BE-CA共晶的粉末X射线衍射峰与物理混合物明显不同,出现了新的特征衍射峰(3.49°、6.15°、7.02°、9.44°、12.25°、14.25°、23.87°、24.69°、26.93°和28.99° 2θ),表明其为不同于BE及CA的新晶体。

Figure5 X-ray powder diffraction patterns of BE,CA,PM and BE-CA cocrystal
3.1.5 扫描电子显微镜(SEM) 由扫描电子显微镜图中可以看出,与本课题组之前研究报道的呈短棒状BE晶体[22](图6-A)不同,所制备的BE-CA共晶(图6-B)呈现长针状。

Figure6 Scanning electron microscope diagram for BE (A) and BE-CA cocrystal (B)
3.2 共晶的体外溶出行为研究
3.2.1 平衡溶解度的测定 由图7可知,由于黄芩素上的酚羟基呈酸性,所以黄芩素和共晶的溶解度均随pH的增加而增加;但在不同pH环境下,共晶的溶解度均显著高于黄芩素。BE及BE-CA共晶在水中的溶解度分别为(16.82±0.52)μg/mL 及(38.11±1.05)μg/mL,共晶在水中的溶解度较黄芩素显著增加。
3.2.2 表观溶出度 BE、PM和BE-CA共晶在不同介质中溶出度的考察见图8,采用溶出效率(Dissolution efficiency,DE)[25-26]对样品在0.01 mol/L盐酸溶液、水和pH 6.8的磷酸盐缓冲液中的溶出进行评价,结果见表1。
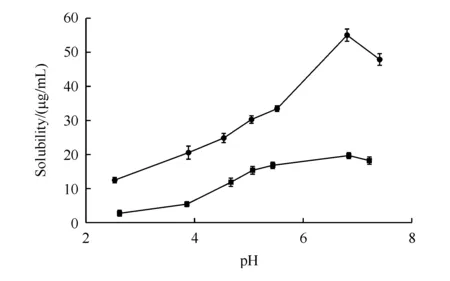

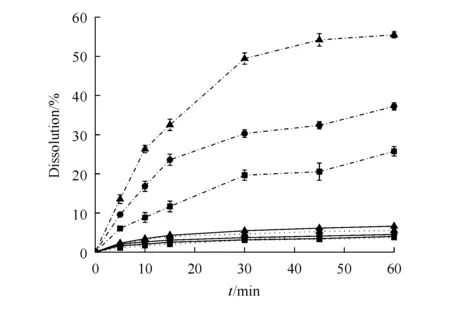
Table1 Dissolution efficiency of different samples
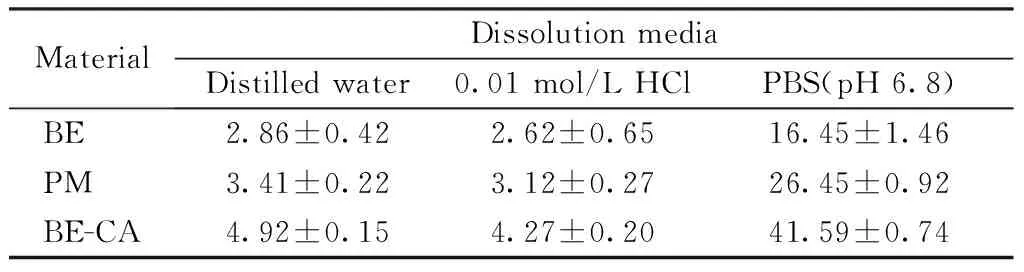
MaterialDissolution mediaDistilled water0.01 mol/L HClPBS(pH 6.8) BE2.86±0.422.62±0.6516.45±1.46PM3.41±0.223.12±0.2726.45±0.92BE-CA4.92±0.154.27±0.2041.59±0.74
DE按如下公式(1)计算:
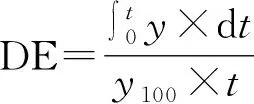
(1)
其中,y为药物在t时刻的溶出度。
结果可知,PM和BE-CA共晶与BE的溶出均具有pH依赖性,pH越高,溶出度越大,且在3种溶出介质中,BE-CA共晶的溶出程度均显著高于BE晶体及PM。在pH 6.8的磷酸盐缓冲液中,BE-CA共晶60 min时的溶出度分别是BE及PM的2.53和1.57倍。
3.2.3 特性溶出速率 BE、PM和BE-CA共晶在不同介质中BE的特性溶出见图9,结果可知在3种不同溶出介质中,共晶中BE的溶出速率均高于BE及物理混合物;经拟合计算特性溶出速率,与单纯BE相比,形成BE-CA共晶后,溶出速率提高了2.5~5倍。
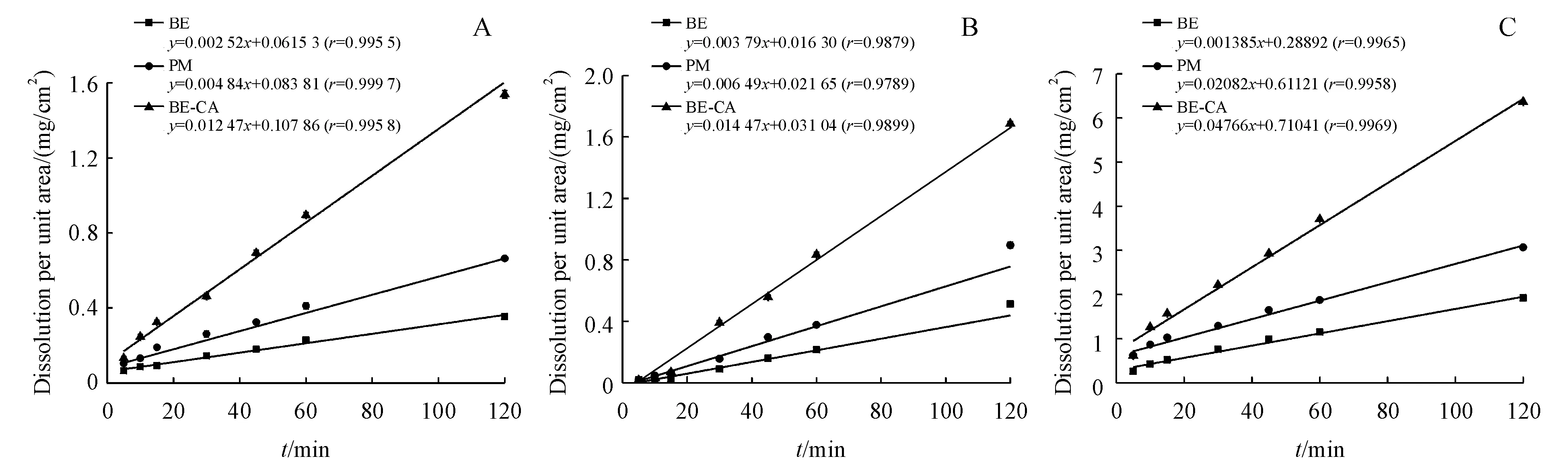
Figure9 Intrinsic dissolution profiles of BE from crystalline BE,PM and BE-CA cocrystal in (A) 0.01 mol/L HCl,(B) water,and (C) pH 6.8 phosphate buffer (n=6)

Solid formsBEtmax /hcmax /(g/mL)AUC0-24 h/(μg·h/mL)BItmax /hcmax /(g/mL)AUC0-24 h/(μg·h/mL)BE0.167±01.67±0.853.05±0.561.50±08.83±1.4274.96±7.92PM0.167±01.85±0.263.12±0.121.50±011.39±0.6380.51±3.77BE-CA0.167±05.85±1.03*7.62±1.35*1.00±0*24.92±2.42*231.44±4.79*
*P<0.05vsBE group
3.3 共晶的体内药代动力学研究
灌胃给予BE、PM及BE-CA共晶后,大鼠体内原性药物BE及活性代谢物黄芩苷(BI)的血药浓度经时曲线如图10所示。给药后大鼠体内活性代谢物BI的血药浓度经时曲线均呈现了双峰现象。该现象的原因是由于胃肠道直接吸收的BE在肠上皮细胞和肝脏中酶的作用下被代谢为BI,形成了BI的第1个血药浓度高峰;之后血液中的BI经肝肠循环,在肠内被酶解生成BE,吸收后的BE在肠上皮细胞及肝脏中酶的作用下又被代谢为BI,即形成BI的第2个血药浓度高峰[27-28]。
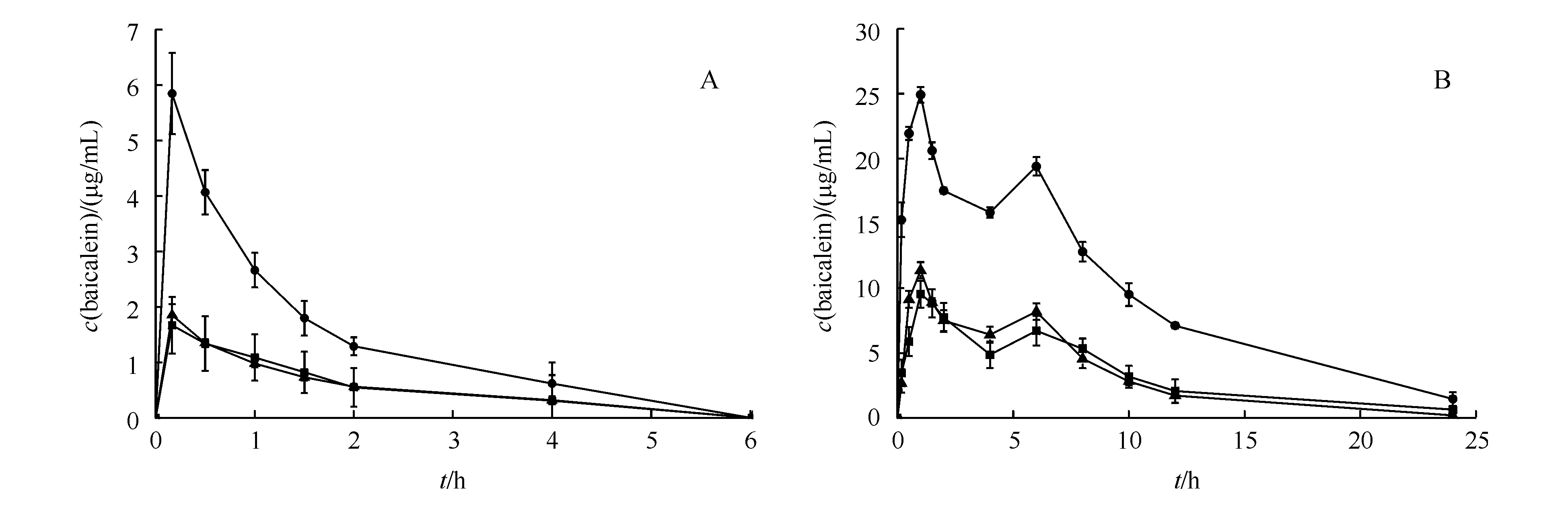
体内药代动力学研究表明,单剂量给药24 h后,与BE晶体组相比,物理混合物PM组的BE及代谢物BI的血药峰浓度(cmax)、达峰时间(tmax)和AUC等药代动力学参数均没有明显变化(表2),说明CA对BE的吸收及代谢影响较小。然而,在给予BE-CA共晶后不仅缩短了BE及BI的达峰时间(tmax),血药峰浓度(cmax)提高了2.8~3.5倍,药时曲线下面积(AUC)提高2.5~3倍,显著提高了黄芩素的吸收。这主要是由于共晶改变了黄芩素原有的结构,提高其溶解度和溶出度,从而提高了其生物利用度。
4 结 论
本实验采用混悬液结晶法制备出长针状BE-CA共晶,根据红外图谱推测,共晶中BE分子4位C=O和5位-OH分别与CA中8位=C-H和9位C=N可能形成了氢键。体外溶出实验和体内药代动力学研究表明,BE-CA共晶不仅有效地改善了BE的溶解度和溶出速率,同时显著提高了BE的体内吸收及其与活性代谢物BI的生物利用度,为BE的进一步开发及临床应用奠定了基础。
猜你喜欢
纺织标准与质量(2022年3期)2022-08-10 09:11:20
承德医学院学报(2022年2期)2022-05-23 13:01:36
模具制造(2019年3期)2019-06-06 02:11:04
中成药(2018年1期)2018-02-02 07:19:55
中成药(2017年10期)2017-11-16 00:50:08
中成药(2017年10期)2017-11-16 00:50:03
含能材料(2017年1期)2017-03-04 15:46:20
含能材料(2017年7期)2017-03-04 11:16:26
特产研究(2016年3期)2016-04-12 07:16:16
当代化工研究(2016年6期)2016-03-20 16:21:48
