
柔性链段的引入对聚甲基丙烯酸三苯基甲酯性能的影响
2018-02-13 06:37孙思佳牛小玲方家义
西安工业大学学报 2018年6期
孙思佳,牛小玲,方家义
(西安工业大学 材料与化工学院,西安 710021)
随着高分子材料科学的迅速发展,聚甲基丙烯酸三苯基甲酯(PTrMA)[1-2]材料的研究、开发与应用已经成为材料研究的热点之一,PTrMA是一种聚丙烯酸酯类[3-4]材料,被运用在飞机座舱罩、挡风玻璃及其他高透明制品上,也可以被运用在工业、仪器仪表、交通运输、医疗卫生以及工艺美术等领域[5-7].
有关PTrMA的研究已经非常广泛,文献[8]成功地合成了具有稳定单向螺旋构象的PTrMA,系统地研究了甲基丙烯酸三苯基甲酯(TrMA)螺旋选择性聚合的过程,并发现了PTrMA对芳香族外消旋化合物具有良好的识别与拆分作用,可以作为高效液相色谱固定相.德国Kulzerr公司以TrMA为主体研制出了一种具有优异的悬浮和增稠性能的粘合剂,其几乎与所有的非离子、阴离子、两性表面活性剂和多种阳离子聚合物配伍[9].文献[10]研究发现TrMA在聚合时,可以迫使共聚单体采取同样立构的螺旋链结构.文献[11]报道PTrMA具有光学活性中心,可以用来有效诱导共聚物的不对称聚合,得到与预聚物同样螺旋的光学活性嵌段共聚物,这种光学活性随组成变化呈现S型曲线[12].
目前,对TrMA材料的报道较少,其大分子链呈无规立构,由于取代基妨碍了大分子的内旋转,使得大分子链有一定的刚性[13-14],致使其玻璃化温度较高,导致TrMA在室温条件下是一种质硬而透明的特殊材料[15].如何改善它的热稳定性,增加它的使用温度范围,这仍然是亟待解决的重要问题.本文通过向TrMA中引入不同比例的甲基丙烯酸甲酯(MMA)柔性链段,影响它的玻璃化转变温度和结晶度,以提高TrMA的热稳定性和加工性,进而改善该材料的缺陷,拓展其应用范围.
1 实验材料的制备及方法
1.1 实验材料的制备
1.1.1 TrMA的制备
将5.6 g (0.02 mol)三苯基氯甲烷和4 mL(0.028 mol) 三乙胺倒入100 mL三口烧瓶中,再加入2 mL (0.024 mol) α-甲基丙烯酸,用30 mL甲苯溶解,80 ℃反应10 h,反应结束有大量白色固体析出,过滤,滤液用冰水洗涤3次,萃取有机相,有机相用无水MgSO4干燥、过滤,母液旋转蒸发去除溶剂,用甲苯/正己烷(1∶1,v/v)试剂进行重结晶,计算产率.
1.1.2 PTrMA的制备
称取3.31 g (0.01 mol) TrMA加入安培瓶中,再量取5 mL甲苯和6.6 mg (0.04 mmol)的偶氮二异丁腈(AIBN)加入烧瓶中,氮气保护下60 ℃反应24 h,反应结束,正己烷使聚合物沉淀析出,离心分离得到聚合物,60 ℃真空干燥24 h,计算产率.
1.1.3 PTrMA-co-MMA的制备
称取3.31 g (0.01 mol)TRMA和0.75 g(7.5 mmol)MMA,5 mL甲苯和6.6 mg(0.04 mmol)的偶氮二异丁腈(AIBN)加入到安培瓶中,氮气保护下60 ℃反应24h,加入大量正己烷得到聚合物沉淀,离心分离得聚合物后,60 ℃真空干燥24 h,计算产率.PTrMA-co-MMA的反应过程如图1所示.
1.2 实验方法
将所得样品进行KBr压片,采用傅里叶变换红外光谱仪(Fourier Transform Infrared Spectrometer,FTIR)进行红外测试, Nicolet8700型,德国耐驰仪器制造公司,范围为4 000~400 cm-1,扫描次数为32,分辨率为4 cm-1;采用核磁共振波谱仪进行核磁共振氢谱(H-Nuclear Magnetic Resonance,1H-NMR)测试, AVANCE400型,瑞士Bruker公司,振动频率为400 MHz,测试温度为25 ℃;采用差示扫描量热仪(Differential Scanning Calorimeter,DSC)进行热力学测试, 204 F1型,德国耐驰公司,循环加热方式温度范围为50~400 ℃;采用热重分析仪进行热重(Thermo Gravimetric,TG)测试,Q5000IR型,美国沃特世科技有限公司,扫描温度范围为25~500 ℃,升温速率为10 ℃·min-1;采用旋转阳极衍射仪进行X射线衍射(X-Ray Diffraction,XRD)测试, DMAX-RB 12 kW型,日本理学公司,测试衍射角范围为3~60°.

图1 PTrMA-co-MMA的反应过程示意图
2 结果与讨论
2.1 TrMA、PTrMA及PTrMA-co-MMA的IR分析
TrMA、PTrMA及PTrMA-co-MMA的红外曲线如图2所示.由图2单体a可知,单体的曲线上比原料的曲线多出一些峰,分别为2 985 cm-1甲基的碳氢键(C—H)伸缩振动,1 724 cm-1羰基的碳氧双键(C=O)伸缩振动,1 633 cm-1乙烯基的碳碳键(C=C)伸缩振动,1 150 cm-1酯基上碳氧键(C—O)伸缩振动这四个峰,根据文献[9]可知,α-甲基丙烯酸在1 594 cm-1、1 549 cm-1、1 487 cm-1、1 444 cm-1和1 395 cm-1是苯环基碳碳键(C=C)伸缩振动,3 000 cm-1附近的三个峰是苯环上的碳氢键(C—H)伸缩振动.说明三苯基氯甲烷和α-甲基丙烯酸已成功发生酯化反应.
由图2均聚物b可知,3 000 cm-1附近上苯环的碳氢键(C—H)伸缩振动峰,1 594 cm-1左右存在几个苯环基结构的碳碳双键(C=C)伸缩振动峰,1 150 cm-1处酯基上碳氧键(C—O)伸缩振动峰依旧存在,而1 633 cm-1处的峰消失了,说明乙烯基的碳碳键(C=C)伸缩振动已消失,即碳碳双键(C=C)结构已经消失,羰基(C=O)特征峰由1 724 cm-1位移到1 734 cm-1,发生了明显的蓝移现象,这是由于均聚导致单体的双键和羰基的共轭结构(C=C—C=O)消失,均聚物的羰基(C=O)频率升高,说明双键断裂形成长链,单体发生聚合.
由图2共聚物c可看出,在1 388 cm-1、1 269 cm-1、1 242 cm-1、1 150 cm-1和989 cm-1处有PMMA中的酯基(-OCH3)特征峰,说明MMA被引入,与TrMA形成共聚物.

图2 单体a、均聚物b和共聚物c的红外光谱图
2.2 TrMA、PTrMA及PTrMA-co-MMA的1H-NMR分析
根据TrMA、PTrMA及PTrMA-co-MMA的核磁分析可知,单体的氢谱在化学位移δ=2.00×10-6处的峰对应丙烯基α碳原子相连的甲基上质子(CH3—C=C).δ=5.62×10-6、δ=6.25×10-6处的峰对应丙烯基β碳原子上质子(—C=CH2)的化学位移.δ=7.00×10-6~7.50×10-6处的群峰对应三苯基苯环上质子(Ar—H)的化学位移.这四组峰的积分面积比为2.99∶1∶1∶15.05,与目标单体TrMA个数比3∶1∶1∶15基本一致,说明单体合成成功.
与单体的氢谱相比,均聚物的氢谱在δ=5.62×10-6、δ=6.25×10-6处无双峰,说明成功发生聚合.δ=1.10×10-6~1.30×10-6的峰群对应聚合物主链上亚甲基的质子(-C-CH2-C-)和侧链甲基的质子(-C(CH3)-C-)的化学位移.δ=7.00×10-6~7.50×10-6的峰群对应三苯基苯环上质子(Ar-H)的化学位移.δ=3.73×10-6对应(CH3-O-)质子的化学位移,这是聚合物与甲醇发生醇解而出现甲氧基的结构.除此之外,图谱中还有四处细小的峰,分别为δ=0.88×10-6,1.27×10-6,2.36×10-6和7.17×10-6.其中δ=0.87×10-6和δ=1.25×10-6为溶剂正己烷的甲基上的质子(-CH3)和亚甲基上质子(-CH2-)的化学位移.而δ=2.36×10-6和δ=7.17×10-6对应溶剂甲苯上甲基的质子(Ar—CH3)和苯环上的质子(Ar—H)的化学位移.考虑到单体聚合相连接时,会出现“头尾相连”(-C-CH2-C-CH2-)和“尾尾相连”(-C-CH2-CH2-C-)的情况,因此,主链上亚甲基的质子会出现不同的位移.但实际图谱上δ=1×10-6~2×10-6的区间,除了δ=1.10×10-6~1.30×10-6和δ=1.56×10-6的宽峰外,再无别的峰,故推测δ=1.56×10-6处的宽峰应是由于不同亚甲基质子的化学位移发生重叠而形成.
从共聚物的氢谱可以看出,在化学位移0.1×10-6~1.86×10-6范围内的五重峰对应于共聚物中MMM,MMT,TMM,TMT和TTT(T∶TrMA∶M∶MMA)三素组序列的α-CH3所处的空间立构的间同、杂同和全同的分布.在化学位移δ=2.01×10-6,δ=2.34×10-6~3.52×10-6和δ=7.23×10-6上,分别为TrMA单元中的侧基中苯环的1位,2-4位及苯基(相联在共聚物端基的催化剂基)的峰.共聚物中的羰基(C-O)峰呈现在化学位移δ=1.75×10-6~1.77×10-6范围,由此可以看出共聚物成功合成.
2.3 PTrMA和PTrMA-co-MMA的DSC及TG分析
2.3.1 PTrMA的DSC及TG分析
PTrMA的TG、DSC曲线如图3所示.由图3(a)可知,PTrMA从130 ℃开始出现质量损失,说明聚合物在主链开始断裂之前已发生侧基的断裂.聚合物侧链的三苯基结构在130~309 ℃这个升温区间逐渐消失,对应DSC曲线里第一个明显的吸热峰.在130~309 ℃区间除了失去三苯基结构,还应伴随着酯基和部分主链的分解.309 ℃之后,失重曲线走势开始变得平缓,直至414 ℃为止,而这段失重曲线应对应残余链段的分解损失,即在DSC曲线上对应第二个明显的吸热峰.
由图3(b)可知,在100 ℃左右开始发生基线的偏移,即对应聚合物的玻璃化转变,说明聚合物的玻璃化转变温度为151 ℃.与热重曲线对比分析可知288 ℃和329 ℃处对应的两个吸热峰为聚合的两个分解峰.
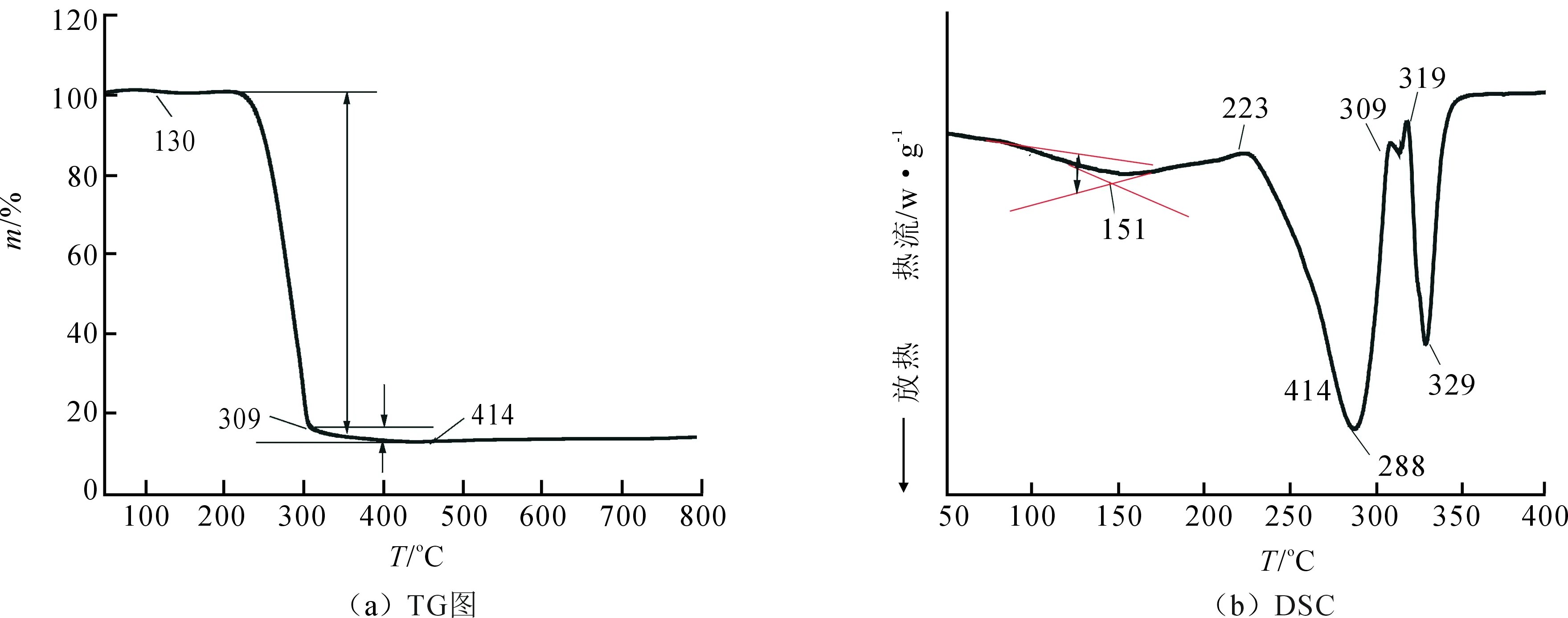
图3 聚合物TG和DSC曲线
2.3.2 不同比例PTrMA-co-MMA的DSC及TG分析
不同比例PTrMA-co-MMA的TG和DSC曲线如图4所示.由图4可知,TrMA和MMA比例为1∶25的共聚物从157 ℃开始出现质量损失,说明先发生侧基的断裂,共聚物侧链的三苯基结构在157~286 ℃升温区间失去了三苯基结构,对应DSC曲线在220 ℃处的第一个明显的吸热峰.而在286~388 ℃这个升温区间质量又开始发生损失,这是共聚后的主链的分解,对应DSC曲线在345 ℃处的第二个明显的吸热峰.在388~440 ℃升温区间内,是其他残余链段的分解损失,对应DSC曲线在446 ℃处的第三个明显的吸热峰,另外两个比例的共聚物与比例为1∶25共聚物具有同样的特征.随着MMA加入量的增加,在TG曲线中起始分解温度分别为138 ℃、148 ℃和157 ℃,在DSC曲线中可以看出吸收峰左移,玻璃化转变温度分别为140 ℃、138 ℃和132 ℃,说明引入MMA可以使聚合物的玻璃化转变温度减小,热稳定性提高.

图4 不同比例共聚物TG和DSC曲线
2.4 不同比例PTrMA-co-MMA的XRD分析
PTrMA和不同比例PTrMA-co-MMA的XRD曲线如图5所示.
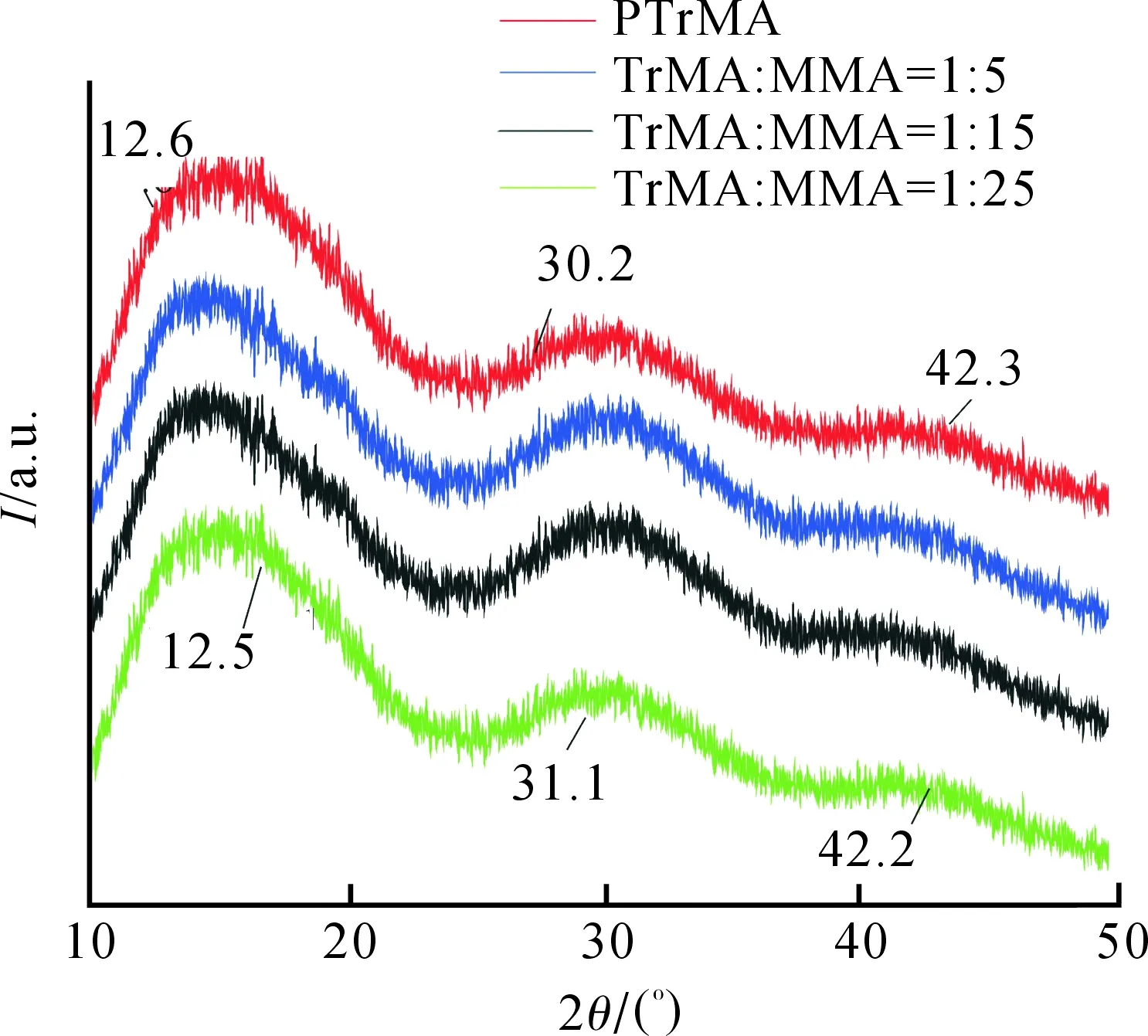
图5 均聚物和不同比例共聚物的XRD曲线
由图5可知,PTrMA在2θ=12.6°,30.2°和42.3°存在三个特殊衍射峰.不同比例PTrMA-co-MMA也出现了与均聚物类似的三个衍射峰,即2θ=12.5°,31.1°和42.2°,因此,可认为共聚物的结晶参数几乎与均聚物完全相同,MMA单体在链段中并不影响原均聚物的结晶结构.随着MMA引入量的增大,衍射峰的面积有递减趋势,证明引入MMA使结晶度降低.
3 结 论
1) 本实验成功地合成了TrMA,以及通过热聚合法制得PTrMA和不同比例的PTrMA-co-MMA.
2) PTrMA的玻璃化转变温度为151 ℃,分解起始温度为130 ℃,通过引入1∶5,1∶15和1:25三个比例的MMA后,共聚物的起始分解温度分别为138 ℃、148 ℃和157 ℃,DSC曲线中的吸收峰出现左移现象,玻璃化转变温度分别为140 ℃、138 ℃和132 ℃,说明引入MMA可以使聚合物的热稳定性提高,玻璃化转变温度减小.
3) PTrMA在2θ=12.6°,30.2°和42.3°存在三个X射线特殊衍射峰,不同比例的PTrMA-co-MMA也出现了与均聚物类似的三个衍射峰,即2θ=12.5°,31.1°和42.2°,并且随着MMA引入量的增大,衍射峰的面积有递减趋势,证明引入MMA并不影响共聚物的结晶结构,但可使结晶度逐渐降低.
猜你喜欢
中学化学(2022年5期)2022-06-17
华南师范大学学报(自然科学版)(2021年4期)2021-08-30
农药科学与管理(2019年8期)2019-11-23
理科考试研究·高中(2019年8期)2019-09-19
江苏农业科学(2018年24期)2018-02-13
吉林农业(2016年8期)2016-05-14
化学教学(2015年11期)2015-12-19
浙江理工大学学报(自然科学版)(2015年7期)2015-03-01
郑州大学学报(医学版)(2015年2期)2015-02-27
中国塑料(2014年4期)2014-10-17
