
肺功能检查预测系统性硬化症相关肺动脉高压的初步研究
2018-02-05 03:44张磊徐建高照猛毛东梅
中国现代医学杂志 2018年4期
张磊,徐建,高照猛,毛东梅
(胜利油田中心医院1.风湿免疫科,2.心血管内科,山东 东营 257034)
系统性硬化症(systemic sclerosis,SSc)是一种原因不明的临床上以局限性或弥漫性皮肤增厚和纤维化为特征的结缔组织病,其最严重的并发症和死亡原因是肺脏受累,如肺间质纤维化(pulmonary interstitial fibrosis,PIF)和肺动脉高压(pulmonary hypertension,PAH),雷玲等[1]研究发现高达53.6%患者合并PIF,而合并PAH的发病率也有10%~30%,但合并PAH患者2年生存率仅为40%~50%[2],故早期诊断并加以处置对于改善患者预后尤为重要。在非创伤性检查操作中,超声心动检查对于发现和诊断PAH有一定优势,但是对于中低程度PAH患者,其有不同程度假阳性及假阴性率,而且超声心动检查对于肺功能预后及PAH进展的预测价值也有限,在心脏超声中发现的肺动脉压增高患者中仅仅有20%左右会进展为严重的PAH[3]。在SSc患者中行肺功能检查可发现不同程度的限制性通气障碍,肺活量减低,肺顺应性降低,气体弥散量减低等,肺功能指标的异常甚至早于临床症状数年。
本研究回顾性分析早期SSc合并PAH患者的相关临床表现、实验室指标、肺功能检查及其相关随访结果,探讨肺功能检查指标的变化对于预测SSc合并PAH患者的肺功能恶化或PAH进展的应用价值。
1 资料与方法
1.1 一般资料
随机选取2010年7月-2014年8月胜利油田中心医院门诊及住院的SSc患者46例。其中,男3例,女43例;年龄30~64岁,平均(44.16±10.12岁);病程1~36个月,平均18个月。所有患者符合1980年美国风湿病学会(ACR)SSc分类诊断标准[4]。
1.1.1 PAH诊断标准 依据2004年欧洲心脏病协会PAH诊断指南,采取无创性心脏彩色多普勒检查(根据三尖瓣反流压差计算):PAH定义为平均肺动脉压静息时>25 mmHg(1 mmHg~0.133 kPa)或运动时>30 mmHg。
1.1.2 PAH严重程度分级 26~40 mmHg为轻度,41~70 mmHg为中度,≥71 mmHg且伴有胸闷、呼吸困难症状或在上述基础上出现右心衰竭为重度。PAH严重程度还可按纽约心脏病协会(NYHA)分级,即Ⅰ、Ⅱ级为轻度,Ⅱ、Ⅲ级为中度,Ⅲ、Ⅳ级为重度[5]。
1.2 排除标准
①非黄色人种;②长期大量吸烟者;③单纯PIF无PAH者;④有慢性肺部疾病者,如慢性支气管炎、慢性阻塞性肺疾病等;⑤严重心血管疾病或先天性心脏病,如主动脉狭窄、房室间隔缺损、扩张性心肌病、二尖瓣狭窄、动脉导管未闭等;⑥肿瘤;⑦胸腹手术病史;⑧急、慢性感染;⑨神经系统疾病。
1.3 病例分组
依据上述诊断标准及排除条件,将入选的SSc患者分为3组:合并PAH组(SSc-PAH组),女性15例,年龄34~62岁,平均(48.33±10.01)岁;合并PAH以及PIF组(SSc-PAH+PIF组),女性13例,男性2例,年龄36~67岁,平均(45.67±11.37)岁;不合并PAH及PIF者为单纯SSc组,女性15例,男性1例,年龄33~61岁,平均(44.67±11.06)岁。
1.4 治疗情况
所有SSc患者均接受强的松、青霉胺、依地酸钙钠(EDTA)等治疗,SSc-PAH组给予吸氧、口服抗凝剂、钙离子拮抗剂治疗,SSc-PAH+PIF组患者在糖皮质激素治疗的基础上还接受6~18个月环磷酰胺(先给予0.6 g/次,1次/2周,连续3个月,然后1.0 g/次,1次/月,连续3个月,再改为1.0 g/次,1次/3月,连续6~12个月)冲击治疗,不能耐受环磷酰胺者则接受霉酚酸酯(40 mg/kg·d)治疗。基础病情缓解后继续应用小剂量泼尼松(<10 mg/d)治疗,是否继续免疫抑制剂维持治疗由其主管医师决定。病情需要及患者经济条件许可可联合靶向治疗(肺血管扩张剂)。轻度PAH者给予免疫抑制剂+PAH基础治疗,中、重度PAH者除上述治疗外还予以靶向治疗。
1.5 方法
1.5.1 标本的采集 受试者空腹8~10 h取肘静脉血7 ml,待测标本最好是新鲜采集的病人血清或血浆,若当时不检测,可将血清或血浆分离出来,置2℃~8℃冷冻保存。若标本中有微粒可低速离心后取上清检测。
1.5.2 肺功能测定方法 肺功能检查采取德国耶格公司肺功能仪。肺总量(total lung capacity,TLC)采用一氧化氮稀释法,用力肺活量(forced vital capacity,FVC)、第1秒用力呼气量采用肺量计测量。肺一氧化碳弥散量测定采用单口呼吸法。入选的全部患者分别在接受治疗前、第24个月进行2次肺功能测量,均采用立位检查,每项指标测量3次,取平均值。其他指标均以实测值占同性别年龄预计值的百分数表示。当肺功能指标小于正 常预计值的80%时认为存在异常,肺功能指标下降15%时定义为肺功能降低。
1.5.3 心脏超声测定方法 根据SKJAERPE和HATLE提出三尖瓣最大反流速度可以算出右室收缩压的原理[6],均由高年资心脏超声专科医师通过计算三尖瓣反流的跨瓣压差估测肺动脉收缩压(pulmonary arterial systolic pressure,PASP)。
1.6 统计学方法
所有数据采用SPSS17.0软件进行分析,计量资料以均数±标准差(±s)表示,采用单因素方差分析,计数资料以例表示,采用Pearson χ2检验,P<0.05为差异有统计学意义。
2 结果
2.1 各组临床及实验室特征比较
SSc-PAH组、SSc-PAH+PIF组雷诺现象、呼吸道症状、指端溃疡、食管钡餐异常、影像学异常的患者例数与SSc组比较,差异有统计学意义(P<0.05),SSc-PAH组、SSc-PAH+PIF组高于SSC组。见表1。
2.2 各组间肺功能指标比较
2.2.1 基线时肺功能情况 基线时SSc-PAH组和SSc-PAH+PIF组的肺功能异常检出率与SSc组比较,差异有统计学意义(P<0.05),SSc-PAH组和SSc-PAH+PIF组高于SSc组;肺一氧化碳弥散量(diffusion capacity for carbon monoxide of lung,DLCO)下降在SSc-PAH组和SSc-PAH+PIF组更常见,FVC下降在SSc-PAH+PIF组更常见,两指标异常率与SSc组比较,差异有统计学意义(P<0.05),SSc-PAH组和SSc-PAH+PIF组高于SSC组;FVC/DLCO值>1.8在SSc-PAH组和SSc-PAH+PIF组中分别为26.7%和46.7%,与SSC组的6.2%比较,差异有统计学意义(P<0.05)。诊断PAH时已有82.5%的患者出现DLCO下降,其中43.3%的DLCO<45%,与SSC组比较差异有统计学意义(P<0.05)。见表 2。
2.2.2 24个月时肺功能比较SSc-PAH组和SSc-PAH+PIF组各项肺功能指标、异常检出率分别与基线时比较,差异有统计学意义(P<0.05);FVC/DLCO值>1.8在SSC-PAH组和SSC-PAH+PIF组中分别为60.0%和80.0%,与SSC组的18.8%比较,差异有统计学意义(P<0.05),同时分别高于基线时各组百分比。见表3。
2.2.3 各组间PAH比较SSC-PAH组在24个月时有3例肺动脉压力由轻度进展为中度;SSC-PAH+PIF组在24个月时有3例肺动脉压力由轻度进展为中度,2例进展为高度。见表4。
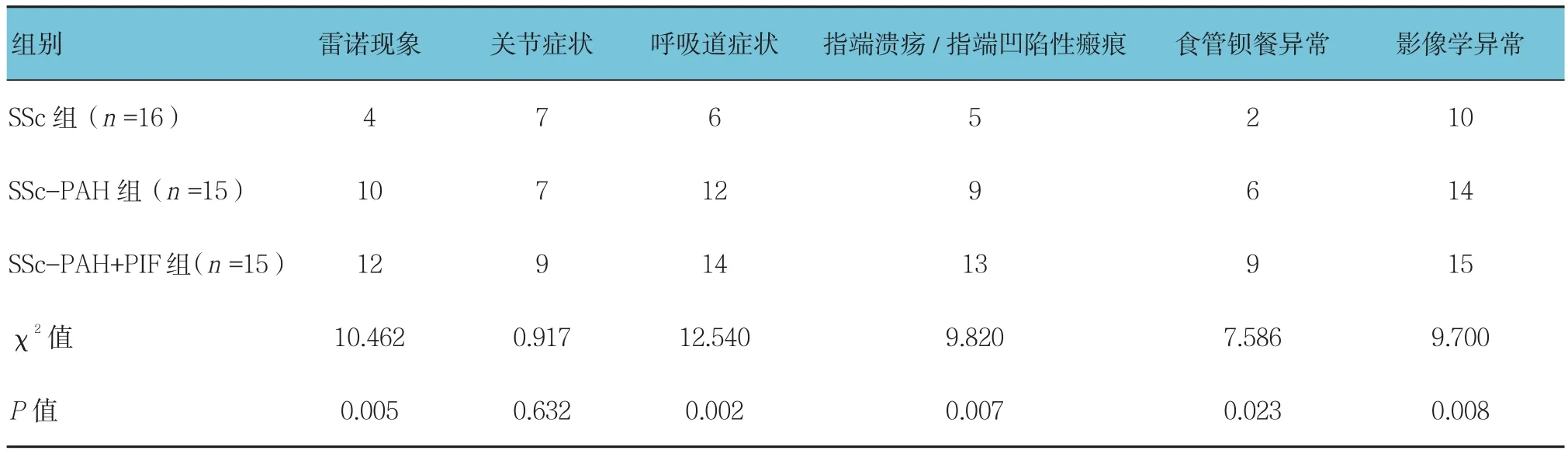
表1 各组临床特征比较 例
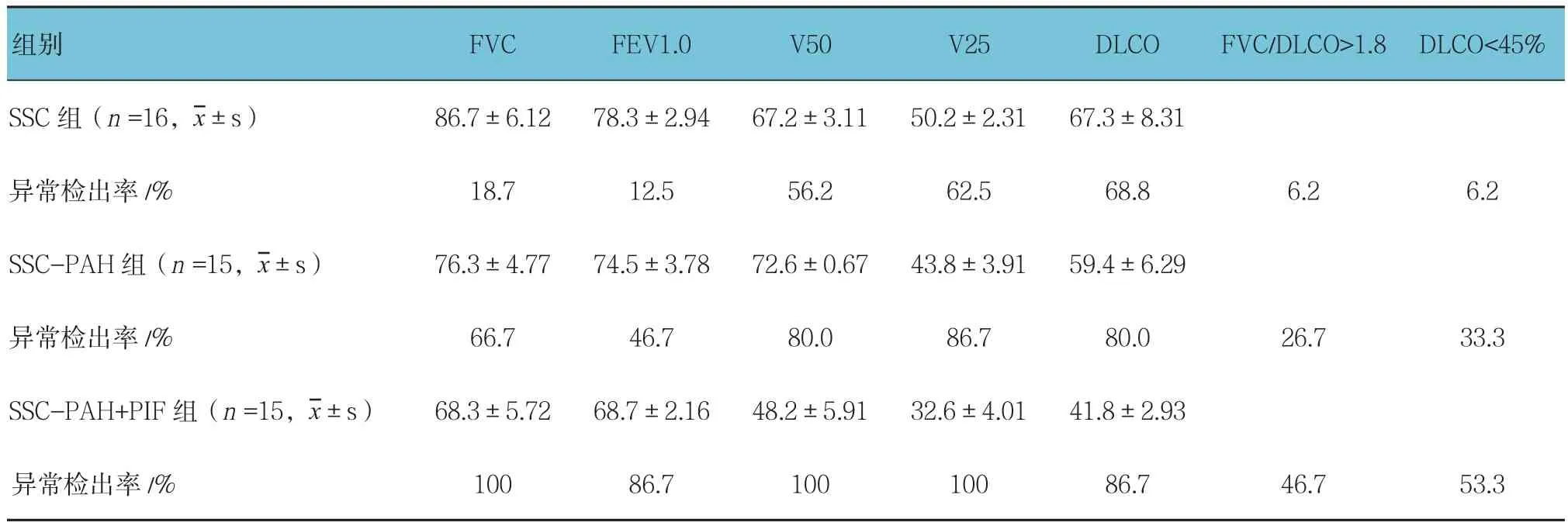
表2 各组基线时肺功能测定结果及异常检出率
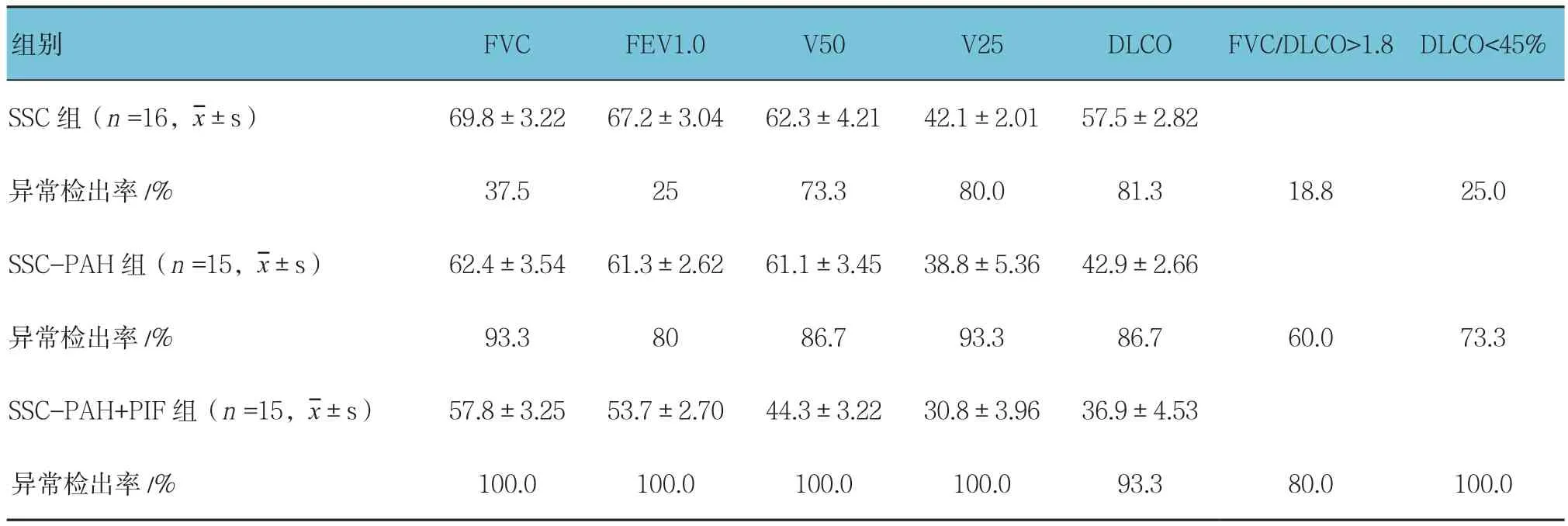
表3 24个月时各组肺功能测定结果及异常检出率
表4 3组患者初诊及24个月时PAH变化比较 (±s)
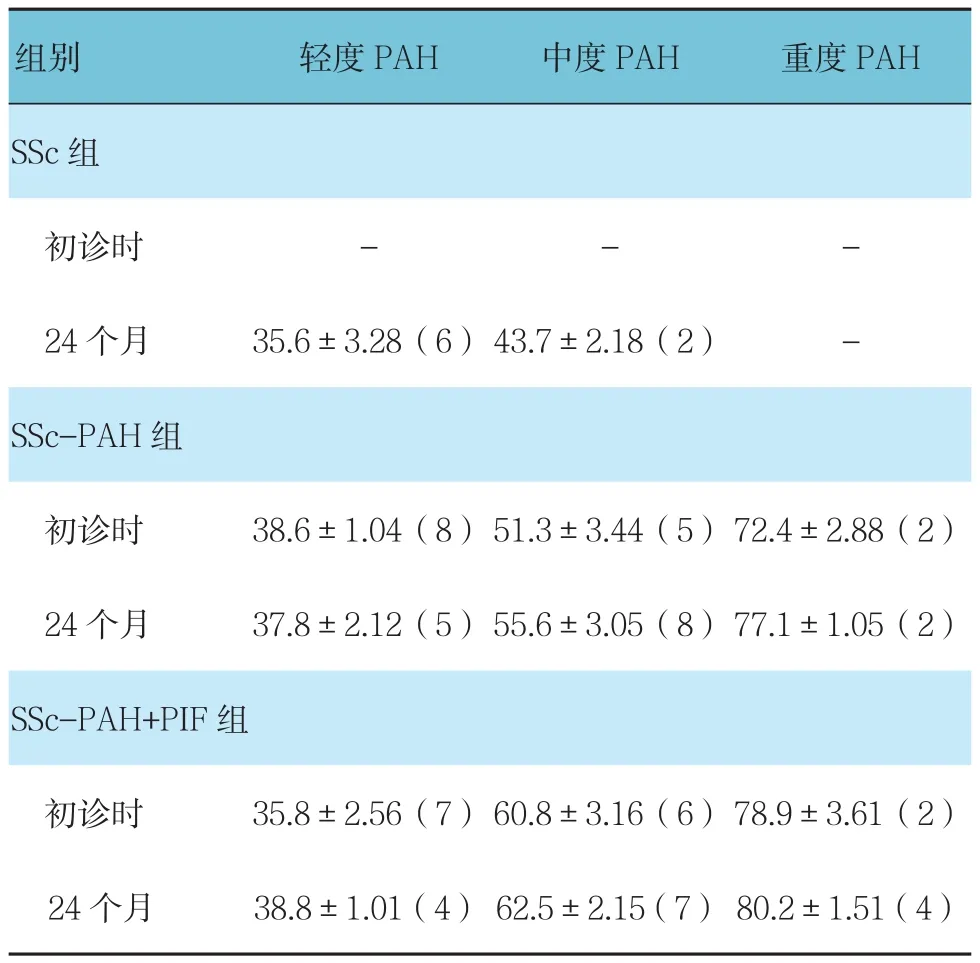
表4 3组患者初诊及24个月时PAH变化比较 (±s)
注:数据后括号内为病例数
组别 轻度PAH 中度PAH 重度PAH SSc组初诊时 - - -24个月 35.6±3.28(6)43.7±2.18(2) -SSc-PAH组初诊时 38.6±1.04(8) 51.3±3.44(5) 72.4±2.88(2)24 个月 37.8±2.12(5) 55.6±3.05(8) 77.1±1.05(2)SSc-PAH+PIF组初诊时 35.8±2.56(7) 60.8±3.16(6) 78.9±3.61(2)
3 讨论
SSc合并PAH的机制复杂,抗内皮细胞抗体、抗成纤维细胞抗体、抗拓扑异构酶Ⅱ-a抗体等自身抗体介导的免疫反应[7-8],血管内皮的损伤、肺血管壁的重塑以及血栓的形成等因素导致肺血管阻力增加,各种细胞因子及生长调节因子,如单核细胞趋化蛋白-1、血小板源性生长因子、生长分化因子15、白细胞介素-6等,均参与了SSc相关PAH的发病[9-10]。
研究发现SSc合并PAH的患者中50%与肺间质病变有关[11],随间质病变日益加重,一方面肺血管床储备减少,特别是肺内小动脉的数量减少,另一方面严重影响气体交换,导致低氧血症,从而导致肺动脉压力逐渐升高,STEEN等[12]认为部分SSc患者的PAH源自于肺间质病变。本研究中患者继发PIF15例,其中轻度PAH者占45.7%,中重度PAH者占53.3%,有PIF患者的肺动脉压力平均值高于无PIF患者。
90%SSc患者出现肺功能异常,且常先于临床症状或影像学改变出现,曾有国外学者统计[13]近50%的SSc患者的胸片检查正常。本研究发现无任何呼吸道症状的SSc患者中有86%影像学出现肺间质改变,限制型通气功能障碍占42.3%,小气道功能障碍占56.8%。但弥散功能下降者却高达91.1%,提示早期患者便可出现限制性通气障碍和弥散功能减低,与蒋明[14]、DOMAGALA[15]观点一致,亦有学者认为SSc肺部受累早期小气道损害为主[16],FEF 25%~75%可以反映小气道病变,具有较好的敏感性和特异性,本研究中发现无呼吸道症状的患者中有56.8%小气道功能异常,有呼吸道症状的患者中有71.3%小气道功能异常,两者比较差异无统计学意义。同时研究发现无呼吸道症状者有2项肺功能指标异常,而有呼吸道症状者则有5项指标异常,并且比无呼吸道症状者有下降的趋势,另外弥散功能异常者中影像学异常占83.2%,弥散功能正常者中有37.6%影像学异常,表明弥散功能损害与影像学异常相关,呼吸道症状可以反映肺功能进展情况,但敏感性较差。
SSc相关肺损害以限制性通气功能障碍、肺顺应性下降、弥散功能减退为特征,主要表现为肺总量、肺活量、FVC、残气量、DLCO下降,其中早期最常见的为DLCO降低。DLCO下降被认为是SSc-PAH的危险因素[17],同时FVC/DLCO比值的变化已被用于SSc患者PAH的风险预测和病情评价[18]。对于SSc相关的孤立性PAH,常有FVC/DLCO比值的增高,源于SSc肺血管损伤,随着血管内膜的增殖,逐渐出现DLCO降低,导致FVC/DLCO比值增高。而合并有PIF时,会出现FVC降低,此时由于肺血管损伤、肺泡滤过膜损伤等造成DLCO不成比例的降低,通常也会出现FVC/DLCO比值增高,即使对于有严重PIF、低氧血症及继发的PAH患者也常常会出现FVC/DLCO比值增高[19],对于SSc合并PAH患者行肺功能检查,无论PAH起因如何,都可依据FVC/DLCO比值可以有效评估病情并一定程度上预测疾病进展。
最后由于本研究病例数及观察期等限制,与目前相关国内外研究出现部分不一致的结果,相信随着将来更多大样本、多中心、前瞻性的研究实施,会为SSc相关PAH的早期诊治提供更为科学的依据。
[1]雷玲, 赵铖, 米村东, 等. 不同结缔组织病肺间质病变的临床特点及治疗效果分析[J]. 实用医学杂志, 2010, 26(13): 2370-2372.
[2]BATTLERW, DAVITT M A, COOPER S M, et al. Prevalence of pulmaonary hypertension in limited and diffuse scleroderma[J].Chest, 1996, 110: 1515-1519.
[3]MACGREGOR A J, CANAVAN R, KNIGHT C, et al. Pulmonary hypertension in systemic sclerosis: risk factors for progression and consequences for survival[J]. Rheumatology (Oxford), 2001, 40:453-459.
[4]中华医学会风湿病学会. 系统性硬化症诊治指南(草案)[J]. 中华风湿病学杂志, 2004, 8(6): 377-379.
[5]British Cardiac Society Guideline and Medical Practice Committee.Recommendations on the management of pulmonary hypertension in clinical practice[J]. Heart, 2001, 86 Suppl 1: 11-13.
[6]SKJAERPE T, HATLE L. Diagnosis and assessment of tricuspid regurgitation with Doppler ultrasound[J]. Echocardiology, 1981,299.
[7]NEGI V S, TRIPATHY N K, MISRA R, et al. Antiendothelial cell antibodies in scleroderma correlate with severe digital ischemia and pulmonary aterial hypertension[J]. J Rheumatol, 1998, 25:462-466.
[8]TORMEY V J, BUNN C C, DENTON C P, et al. Antifibrillarin antibodies in scleroderma correlate with severe digital ischemia and pulmonary arterial hypertension[J]. J Rheumatol, 1998, 25:462-466.
[9]同生, 毛毅敏, 孙玉霞, 等. 白介素-6和内皮素-l在慢性阻塞性肺疾病合并肺动脉高压患者中的表达[J]. 实用学杂志, 2012,28(12): 1977-1979.
[10]CHRISTMAN B W, MCPHERSON C D, NEWMAN J H, et al. An imbalance between the exeretion of thromboxane and prostacyclin metabolites in pulmonary hypertension[J]. N Engl J Med, 1992, 327: 70-75.
[11]TRAD S, AMOURA Z, BEIGELMAN C, et a1. Pulmonary arterial hypertension is a major mortality factor in diffuse systemic sclerosis, independent of interstitial lung disease[J]. Arthritis Rheum, 2006, 54: 184-191.
[12]STEEN V. Predictors of end stage lung disease in systemic sclerosis[J]. Ann Rheunl Dis, 2003, 62: 97-99.
[13]SPAGNOLATTI L, ZOIA M C, VOLPINI E, et al. Pulmonary function in patients with systemic sclerosis[J]. Monaldi Arch Chest Dis, 1997, 52(1): 4-8.
[14]蒋明, 风湿病学[M]. 北京: 北京科技出版社, 1995: 625.
[15]DOMAGALA L J, WOSER G, DOBOSZYNSKA A, et al.Interstitial lung disease insystemic sclerosis: comparison of BALF lymphocyte phenotype and DLCO impairment[J]. Respir Med, 1998, 11: 1295-1301.
[16]GUTTADANRIA M. Pulmonary function in scleroderma[J].Rheum, 1977, 20: 107.
[17]STEEN V, CHOU M, SHANMUGAM V, et a1. Exerciseinduced pulmonary arterial hypertension inpatients with systemic sclerosis[J]. Chest, 2008, 134: 146-151.
[18]CELEBI-SOZENER Z, KARABIYIKOGLU G, DUZGUN N.Evaluation of the functional parameters in scleoderma cases with pulmonary involvement[J]. Tuberk Toraks, 20l0, 58: 235-241.
[19]STEEN V D, LUCAS M, FERTIG N, et al. Pulmonary arterial hypertension and severe pulmonary fi brosis in systemic sclerosis patients with a nucleolar antibody[J]. J Rheumatol, 2007, 34(11):2230-2235.
猜你喜欢
中国临床医学影像杂志(2022年5期)2022-07-26
昆明医科大学学报(2021年6期)2021-07-31
世界最新医学信息文摘(2021年12期)2021-06-09
世界最新医学信息文摘(2021年12期)2021-06-09
现代临床医学(2021年2期)2021-03-29
中国生殖健康(2020年8期)2021-01-18
中国生殖健康(2020年7期)2021-01-18
心肺血管病杂志(2020年5期)2021-01-14
中国临床医学影像杂志(2019年1期)2019-04-25
中国体外循环杂志(2015年3期)2015-12-08
