
番茄U6启动子的克隆及CRISPR/Cas9基因编辑体系的建立
2018-02-04 03:03蒲艳刘超李继洋阿尔祖古丽塔什胡燕刘晓东
中国农业科学 2018年2期
蒲艳,刘超,李继洋,阿尔祖古丽·塔什,胡燕,刘晓东
番茄启动子的克隆及CRISPR/Cas9基因编辑体系的建立
蒲艳,刘超,李继洋,阿尔祖古丽·塔什,胡燕,刘晓东
(新疆农业大学农学院/新疆农业大学农业生物技术重点实验室, 乌鲁木齐 830052)
【目的】从番茄中克隆高效转录的启动子,构建CRISPR/Cas9基因编辑载体,并在番茄中建立CRISPR/Cas9系统,为番茄功能基因组学和分子育种研究提供技术基础。【方法】采用PCR方法从‘中蔬四号’番茄品种中克隆4种启动子,利用Transfer PCR方法分别对4个启动子进行两种不同长度的截短,分别构建8个截短的启动子驱动的植物融合表达载体。利用农杆菌瞬时转化法分别转染番茄叶片,通过染色筛选出在番茄叶片中转录活性较高的-2启动子。采用DNA重组技术构建以-2为启动子驱动sgRNA,以番茄白粉病相关基因和为靶序列的CRISPR/Cas9基因组编辑载体。载体构建成功后,采用PEG法转化番茄原生质体,提取基因组DNA,采用酶切/PCR法分析内源基因突变情况;采用测序法分析内源基因突变的类型。利用突变位点频率分布图来验证番茄内源启动子在番茄CRISPR/Cas9系统中的有效性。【结果】经过两轮PCR,共获得4种8个不同长度的番茄启动子,其长度分别是452、202、448、206、433、190、448和218 bp,启动子序列比对分析发现番茄启动子与拟南芥启动子一样,也含有比较保守的两个元件,USE和TATA框。成功构建了8个启动子分别驱动的植物融合表达载体。番茄叶片染色结果显示转化后的番茄叶片均被染成蓝色,表明克隆的番茄8个启动子均具有转录活性。选择-2P4为启动子驱动sgRNA,成功构建番茄白粉病相关基因和为靶序列的CRISPR/Cas9基因组编辑载体,验证结果表明番茄内源启动子-2P4能有效地驱动sgRNA的转录,并成功实现对番茄内源基因的编辑。内源基因突变的类型都为碱基替换,突变热点仅存在于内源基因靶序列区。【结论】成功克隆了4种在番茄叶片中高效转录的启动子;基于-2启动子的CRISPR/Cas9基因组编辑载体,在番茄中成功实现对内源基因的编辑。
CRISPR/Cas9;启动子;克隆;番茄;基因编辑
0 引言
【研究意义】2012年由14个国家共同完成了番茄全基因组测序工作,预测番茄共有3.4×104—3.5×104个编码蛋白的基因[1],然而绝大多数基因的功能并不清楚。基因编辑技术是近几年兴起的一项能对基因组完成精确修饰的技术,可完成基因定点碱基替换、敲入或小片段的缺失等,从而可为番茄基因功能的研究提供优良的突变体遗传材料,为今后番茄功能基因组学和分子育种方面的研究提供强有力的技术基础。【前人研究进展】目前常见的基因编辑技术主要有锌指核酸酶(zinc-finger nucleases,ZFNs)[2]、TALEN核酸酶(transcription activator like effector nucleases,TALENs)[3]和CRISPR/Cas(规律成簇间隔短回文重复序列,clustered regularly interspaced short palindromic repeats/CRISPR-associated protein)[4]3种技术体系。CRISPR/Cas9系统于2013年兴起,因其操作简单、效率高、脱靶效率低等特点受到了全世界的广泛研究。TypeII型CRISPR/Cas系统是由一个单链向导RNA(single guide RNA,sgRNA)和一个具有靶向识别sgRNA的核酸酶Cas9组成,这两种成分共同介导了靶位点的切割,进而引起靶位点的突变,它是目前应用最为广泛的一个基因编辑系统,目前已成功运用于多种植物体中[5-7]。RNA是一种小的非编码RNA,广泛参与mRNA前体的剪接成熟,其对应的启动子能驱动sgDNA的转录并且是CRISPR-Cas9系统中重要的元件之一[8-12],在真核生物中,启动子发挥其转录功能的两个重要元件是USE和TATA框[13]。刘玟彬等[14]用拟南芥启动子驱动两个sgRNA,成功实现了拟南芥5序列内大片段的缺失。XING等[15]运用拟南芥启动子在玉米和拟南芥中成功实现了内源基因的编辑。LI等[16]克隆拟南芥启动子驱动sgRNA,成功在拟南芥中实现了靶基因和的突变。【本研究切入点】虽然已报道许多不同物种的启动子,在CRISPR-Cas9系统中成功得到了应用,但同样的启动子在亲缘关系较远物种中不一定适用,到目前为止,适用于番茄的内源启动子并无研究报道。生物信息学分析显示,番茄中至少含有10个启动子,这些启动子是否都具有转录功能并不清楚。【拟解决的关键问题】克隆具有高效转录活性的番茄内源启动子并构建相应的CRISPR/Cas9编辑载体,通过瞬时转化番茄原生质体来实现对番茄内源基因的编辑,在番茄中建立有效的CRISPR/Cas9基因编辑技术体系。
1 材料与方法
试验于2016—2017年在新疆农业大学农业生物技术重点实验室进行。
1.1 试验材料
试验所用载体质粒-5P2::、农杆菌GV3101、番茄‘中蔬四号’均由新疆农业大学农学院实验室保存。限制性内切酶购于Thermo公司;EasyPure植物基因组DNA提取试剂盒、T4 DNA连接酶、Blunt Zero载体、Trans5α感受态细胞、1 kb Plus DNA Ladder等均购自于北京全式金生物技术公司;Phusion超保真DNA聚合酶购于北京NEB公司。测序及引物合成均由上海杰李生物科技有限公司完成。
1.2 番茄不同SlU6-2P、SlU6-3P、SlU6-4P、SlU6-9P启动子的克隆
将番茄()品种‘中蔬四号’播种于含蛭石与营养土的土壤中,取生长3—4周的幼苗叶片,采用植物基因组DNA提取试剂盒提取番茄基因组DNA,用保守的RNA序列在番茄基因组数据库(http://www.phytozome.net/)中搜索候选RNA对应的启动子序列,并设计PCR扩增引物(表1)。本所采用两轮PCR的方法,第1轮PCR采用Phusion超保真DNA聚合酶进行扩增,反应程序为94℃ 4 min;98℃ 30 s,58℃ 30 s,72℃150 s,35个循环;72℃ 8 min;反应体系为ddH2O 11 μL、5×Phusion buffer 4 μL、2.5 mmol∙L-1dNTP 1.6 μL、上下游引物各1 μL、DNA模板1 μL、Phusion超保真DNA聚合酶0.4 μL,共20 μL体系。获得的启动子片段分别命名为-2P、-3P、-4P和-9P,用1%琼脂糖凝胶电泳检测正确后,Quick Gel Extraction Kit胶回收试剂盒回收并纯化目的DNA片段。回收完成后与平端载体B-Zero连接、转化,对重组质粒用Ⅰ进行酶切鉴定,正确的质粒进行测序,随后用MegAlign软件对测序的质粒进行序列比对分析,第1轮PCR扩增产物测序正确的质粒用作模板再进行第2轮PCR。

表1 本研究中使用的引物序列
下划线为受体质粒部分碱基序列
Underlined sequences are the part of receptor plasmid partial nucleotide sequence
第二轮采用Transfer PCR[17]的方法,使用Phusion超保真DNA聚合酶进行扩增,克隆不同截短的-2P、-3P、-4P、-9P启动子。先根据第一轮测序正确的4个启动子序列和-5P2::载体序列各设计2对引物(表1)。以-5P2::的质粒为受体质粒,-2P、-3P、-4P、-9P启动子的质粒为供体质粒进行Transfer PCR,反应程序为95℃预变性1 min;95℃变性30 s,60℃退火1 min,72℃延伸90 s,13个循环;95℃变性30 s,67℃退火1 min,72℃延伸4 min,20个循环;72℃延伸8 min。反应体系为ddH2O 32.7 μL、5×Phusion buffer 10 μL、2.5 mmol∙L-1dNTP 2.5 μL、上下游引物各1 μL、供体质粒和受体质粒模板各1 μL、Phusion超保真DNA聚合酶0.8 μL,共50 μL体系。对琼脂糖凝胶电泳检测正确的Transfer PCR产物进行Ⅰ酶切处理,酶切产物转化Trans5α感受态细胞,用HⅠ和Ⅰ酶切鉴定正确的质粒进行测序,用MegAlign软件进行序列比对分析,正确的质粒命名为T-Ps。
1.3 SlU6启动子序列分析
运用DNAMAN软件对拟南芥及已克隆的番茄候选的启动子序列进行比对分析。
1.4 不同截短SlU6Ps::GUS-sgRNA-P1300融合表达载体的构建
用Ⅲ和Ⅰ双酶切番茄启动子不同截短的T--2Ps、T--3Ps、T--4Ps、T--9Ps质粒以及植物表达载体pCAMBIA 1300,分别回收目标片段并用T4连接酶连接、转化Trans5α感受态细胞,对重组质粒用Ⅲ和Ⅰ进行酶切鉴定,鉴定正确的质粒命名为Ps::-sgRNA-P1300,转入农杆菌感受态GV3101,28℃倒置培养2 d后,挑取克隆于LB液体培养基(Kan 50 μg·mL-1,Rif 25 μg·mL-1)培养至对数生长期,然后提取质粒再次用Ⅲ和Ⅰ酶切鉴定,鉴定正确的质粒和农杆菌菌液置于-20℃保存。番茄不同Ps::-sgRNA-P1300融合表达载体示意图见图1。
1.5 不同截短SlU6Ps::GUS在番茄叶片中的瞬时表达分析
将构建好的番茄不同-Ps::GUS-sgRNA- P1300 融合表达载体以及阳性对照pCAMBIA1304,阴性对照pCAMBIA1300植物表达载体的农杆菌分别按照1﹕100比例接种于LB培养基(Kan 50 µg·mL-1,Rif 25 µg·mL-1)中活化,28℃,180 r/min摇菌至菌液OD600=0.6—1.2,分别吸取不同体积的活化菌液接种到5 mL LB培养基(含50 µg·mL-1Kan和25 µg·mL-1Rif)中,使每个农杆菌起始菌液OD值相同,再次28℃,180 r/min摇菌至所有菌液的OD600= 1.5时,12 000×离心5 min收集菌体,弃上清液,重悬于Buffer(50 mmol∙L-1Mgcl2、200 mmol∙L-1MES、20 mmol∙L-1乙酰丁香酮)至1 mL,室温放置3 h后分别注射到生长2—3周的番茄子叶中,注射完成后每种样品做好标记,进行过夜暗培养。用剪刀分别剪下暗培养过夜后的番茄叶片,浸泡在100 µl GUS染液中(0.5 mol·L-1磷酸缓冲液,pH 7.0;0.5 mol·L-1EDTA;pH 8.0;10% Triton X-100;20 mmol·L-1X-Gluc),并在-0.05 MPa下抽真空30 min。每种农杆菌转化进行技术重复3次,生物学重复2次。
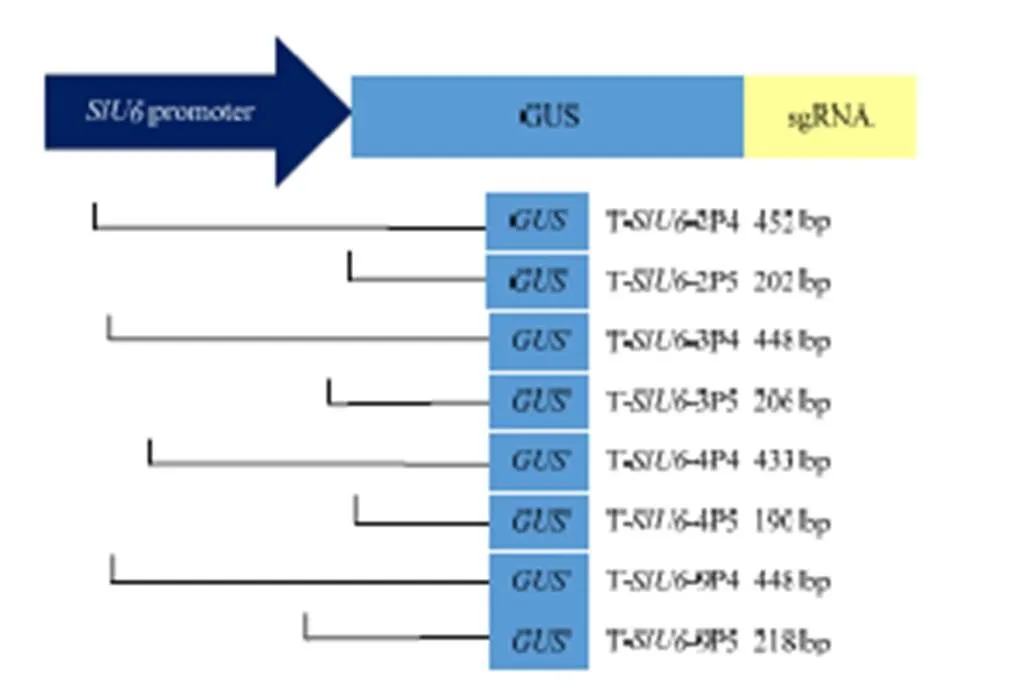
图1 番茄不同截短SlU6启动子驱动GUS的融合表达载体构建
将抽完真空后的番茄叶片置于37℃,180 r/min振荡染色6—7 h,吸尽GUS染液。最后加入300 µl 95%乙醇进行脱色处理,脱色时,每3—4 h更换一次脱色液,直至叶片绿色褪去,最后将脱色后的叶片置于体式显微镜下观察并拍照,根据染色情况筛选出在番茄叶片中高效转录的启动子。
1.6 番茄CRISPR/Cas9载体构建
采用染色较深的启动子构建番茄CRISPR/Cas9基因编辑载体。根据网上公布的番茄基因组数据库,以白粉病相关基因和设计引物进行克隆测序,获得‘中蔬四号’番茄品种和的基因组序列,每个基因设计3个靶序列,并分别构建以和为靶位点的6个sgRNA(表1)。靶序列设计在PAM(5′-NGG-3′)(the protospacer-adjacent motif)位点前,长度为24个碱基,并在PAM位点5′上游的切割位点处为限制性核酸内切酶识别位点。靶序列经过退火(95℃每5 s降低0.5℃至20℃),通过T4连接酶连接过夜,插入到经Ⅰ酶切的T--2p4载体相应位置上,转化-5α感受态细胞,挑取单克隆测序鉴定带有和靶序列的sgRNA表达载体。最终6个带有靶位点的sgRNA载体测序正确后用Ⅰ和Ⅰ酶切,同时酶切Cas9载体后回收目的条带,T4连接酶连接过夜,转化-5α感受态细胞,酶切鉴定后通过琼脂糖凝胶电泳检测结果与预期目的片段大小一致,表明以-2P4启动子驱动带有和靶序列的番茄CRISPR/Cas9载体构建成功。
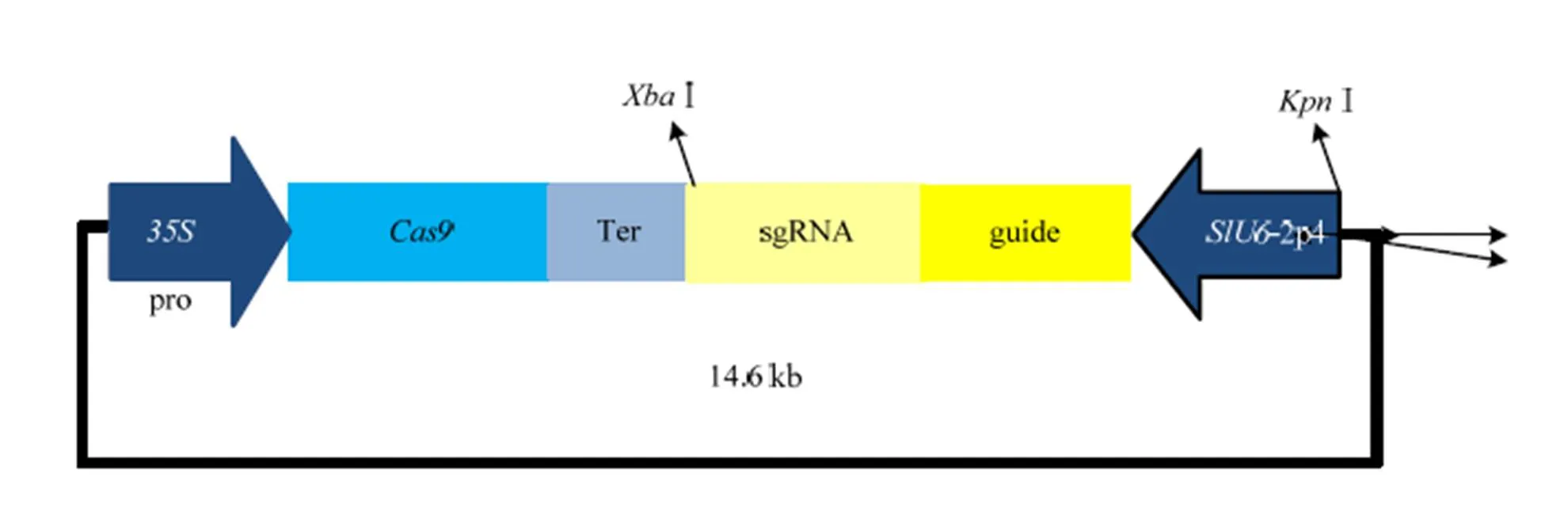
图2 sgRNA和Cas9共表达载体的构建
1.7 番茄原生质体的制备及转化
将‘中蔬四号’番茄品种播种于含蛭石与营养土的土壤中,选取生长在短日照(12 h光照和12 h黑暗)条件下,大约2—3周的番茄子叶。准备两种黏度不同的胶带,先用黏性较强的一面固定于叶片的正面,将黏性较弱的胶带粘住背面,用手轻轻按压,受力均匀,之后缓慢撕下黏性较弱的胶带,下表皮会随着胶带而被撕去,之后叶肉细胞将裸露出来,将这一面叶片放入纤维素酶溶解液中(1.25% celluase R10,0.3% macerozyme R10,0.4 mol∙L-1mannitol,20 mmol∙L-1KCl,20 mmol∙L-1MES,55℃,10 min)酶解,后续试验按文献[18]进行。
在番茄原生质体转化前对带有靶序列的CRISPR/ Cas9载体的核心区域进行PCR扩增,引物见表1(TVV),扩增片段覆盖有或靶序列的-2p4::sgRNA及35S-Cas9-ter两个核心元件,琼脂糖凝胶电泳检测正确后采用酚氯仿抽提和酒精沉淀法对其产物加以简单纯化[19]。番茄原生质体的转化参见文献[18]进行。
1.8 番茄EDR1和MLO1靶序列突变位点检测
将转化后孵育过夜的番茄原生质体用植物基因组DNA提取试剂盒提取番茄基因组DNA,采用基因组DNA先酶切后PCR(Restriction Enzyme digestion/PCR,RE/PCR)的方法[16]对靶序列内突变位点进行检测,引物见表1(Test系列),回收初步检测出突变的PCR产物,通过克隆测序结果比对分析番茄和靶序列内的突变。
1.9 靶基因内不同区域突变频率的计算
为了排除碱基突变是PCR过程中引入随机突变的可能性,对酶切前的番茄基因组DNA进行PCR扩增,引物见表1(Test系列)。PCR产物克隆,选取100个阳性克隆进行测序。以40个碱基为一个计算单元,对包括靶序列(263—286 bp)在内的600 bp的PCR扩增区域内的突变进行频率计算,并对突变频率分布结果进行分析。
2 结果
2.1 SlU6-2P、SlU6-3P、SlU6-4P、SlU6-9P启动子第1轮扩增产物鉴定
从番茄‘中蔬四号’中分别成功扩增出-2P、-3P、-4P、-9P启动子片段,长度分别为1 347、1 385、1 258和1 253 bp。克隆后对酶切鉴定正确的质粒进行测序,结果表明正确(图略)。
2.2 不同截短 T-SlU6扩增产物鉴定
以第1轮测序正确的-2P、-3P、-4P、-9P启动子的质粒为供体质粒,-5P2::的质粒为受体质粒进行Transfer PCR,最终将受体质粒-5P2::中的棉花启动子片段分别置换为番茄的4种8个启动子片段。图3中A和B分别为T--2P4、T-2P5、T-3P4、T--3P5、T--4P4、T--4P5、T-9P4和T--9P5 Transfer PCR的电泳检测图,其启动子截短片段大小依次为452、202、448、206、433、190、448和218 bp。用Ⅲ和Ⅰ双酶切各截短启动子的质粒,结果与预测片段大小一致(图略)。
2.3 不同截短SlU6-Ps::GUS-sgRNA-P1300融合表达载体构建
将测序正确的不同截短Ps::-sgRNA的重组基因片段连入植物表达载体pCAMBIA1300,分别用d Ⅲ和HⅠ分别酶切植物表达载体pCAMBIA1300和8个构建成功的不同截短Ps::-sgRNA融合表达载体,回收目标片段后进行连接,转化,用HⅠ和Ⅰ分别对不同Ps::-sgRNA-P1300重组质粒进行酶切鉴定,2P4::-sgRNA-P1300产生 630 bp和11.2 kb两条带;2P5::-sgRNA-P1300产生380 bp和11.2 Kb两条带;3P5::-sgRNA-P1300 产生390 bp和11.2 kb两条带;4P4::-sgRNA-P1300 产生510 bp和11.2 kb两条带;4P5::-sgRNA- P1300产生370 bp和11.2 kb两条带;9P4::- sgRNA-P1300产生433 bp和11.2 kb两条带;9P5::-sgRNA-P1300 产生400 bp和11.2 kb两条带,以上检测结果均与预测片段大小相符(图4)。
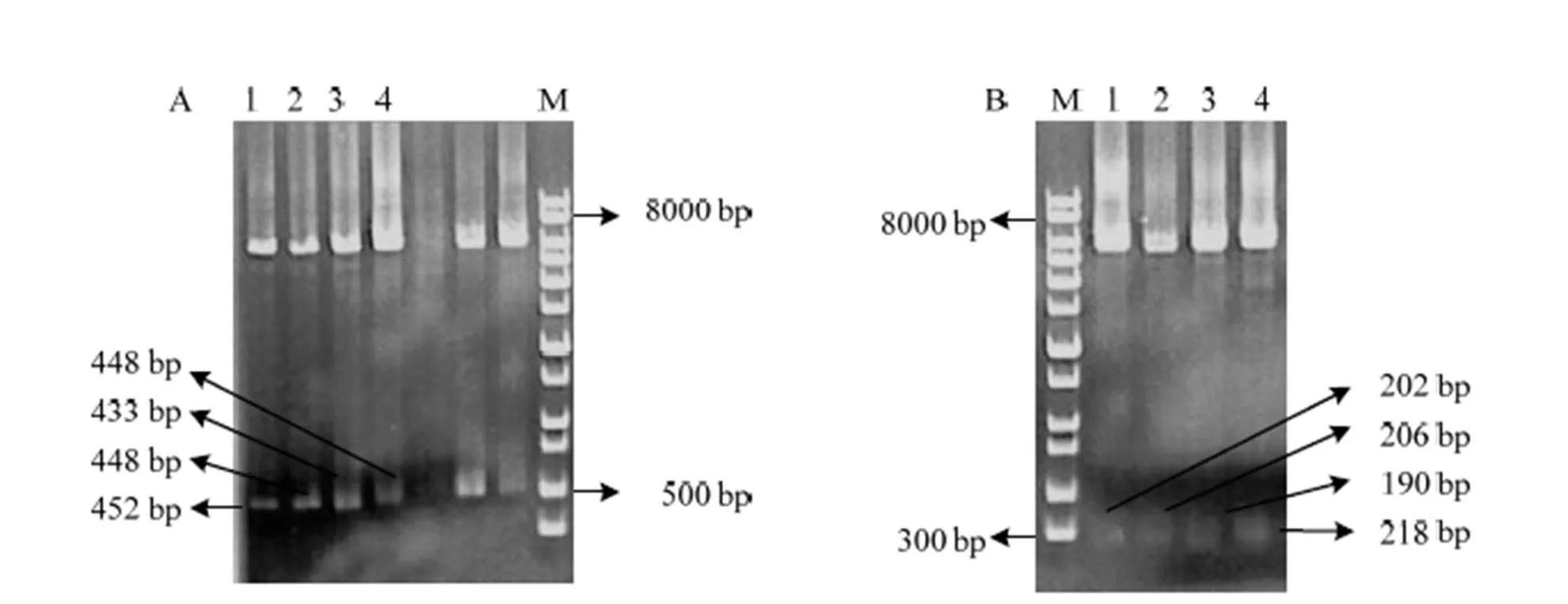
M: 1 kb Plus DNA标准分子量 1 kb Plus DNA marker; A: 1: SlU6-2P4::GUS-sgRNA; 2: SlU6-3P4::GUS-sgRNA; 3: SlU6 4P4::GUS-sgRNA; 4: SlU6 9P4::GUS-sgRNA; B: 1: SlU6-2P5::GUS-sgRNA; 2: SlU6-3P5::GUS-sgRNA; 3: SlU6 4P5::GUS-sgRNA; 4: SlU6 9P5::GUS-sgRNA
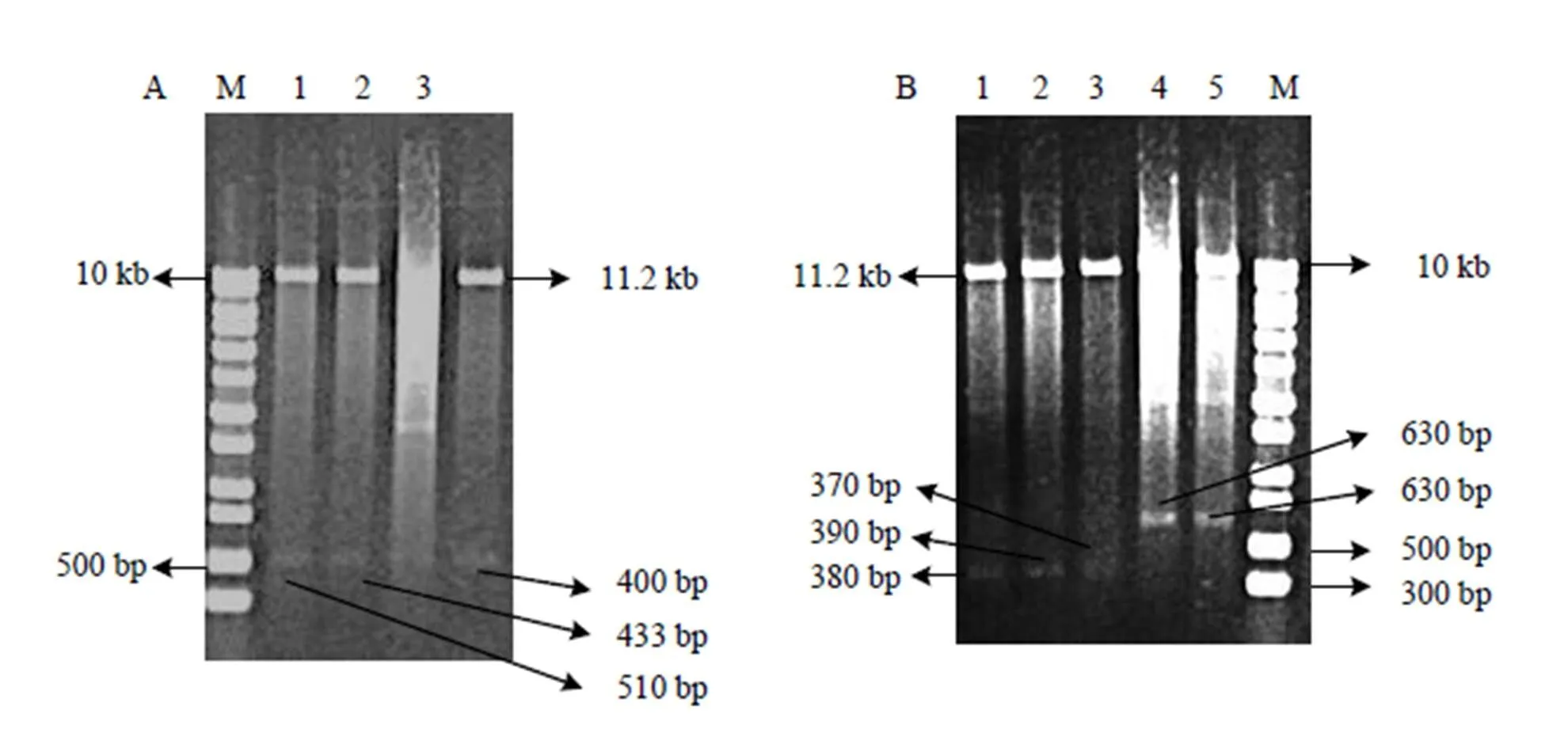
M: 1 Kb Plus DNA标准分子量1 kb Plus DNA marker; A: 1: SlU6-4P4::GUS-sgRNA-P1300; 2: SlU6-9P4::GUS-sgRNA-P1300; 3: SlU6 9P5::GUS- sgRNA-P1300;B: 1: SlU6-2P5::GUS-sgRNA-P1300; 2: SlU6-3P5::GUS-sgRNA-P1300; 3: SlU6 4P5::GUS-sgRNA-P1300; 4: SlU6 2P4::GUS-sgRNA-P1300; 5: SlU6 3P4::GUS-sgRNA-P1300
2.4 SlU6启动子序列分析
对已克隆的4个候选番茄启动子和拟南芥启动子序列进行比对分析,发现番茄启动子序列也非常保守,含有不同于Pol II RNA聚合酶启动子位点的TATA box及USE(upstream sequence element)元件5′-TCCCACATCG-3′,并且这两个元件之间的距离也比较固定(图5)。
2.5 SlU6启动子转录活性测定
将构建好的不同截短Ps::-sgRNA- P1300,阳性对照pCAMBIA 1304及阴性对照pCAMBIA 1300通过农杆菌介导法瞬时转化番茄幼苗叶片。通过GUS组织化学染色分析发现,农杆菌成功侵染后的番茄叶片均被染成蓝色,表明克隆的8种启动子均能驱动报告基因的表达(图6)。
2.6 CRISPR/Cas9载体构建
以番茄-2P4为启动子,构建以白粉病相关基因1和序列内共6个靶位点的sgRNA,测序正确后用Ⅰ和Ⅰ酶切6个带有靶位点的sgRNA载体和Cas9载体后进行连接转化,最终用同样的酶切鉴定挑取的单克隆,通过琼脂糖凝胶电泳检测与预期目的条带大小一致(图7)
2.7 CRISPR-Cas9介导的番茄EDR1和MLO1靶序列突变位点及突变率检测
通过基因组DNA酶切后PCR的方法对靶序列突变位点进行检测,靶区域限制性内切酶识别位点突变后,限制性内切酶就不能进行切割,则PCR扩增出预期产物,而未编辑突变的野生型对照则基本无PCR产物(图8-A、B)。对PCR产物进行回收、克隆和测序。通过序列比对分析发现,内靶位点发生个别碱基的替换(图8-B)。在试验中未检测到所有靶位点和SapⅠ靶位点的突变。对靶基因包括靶序列(263—286 bp)在内的600 bp扩增区域内的突变频率进行计算,结果发现在靶序列处明显出现一个突变峰值,其突变频率明显高于其他区域(图8-D)。表明靶序列区碱基的突变确实是CRISPR/Cas9系统基因编辑的结果。
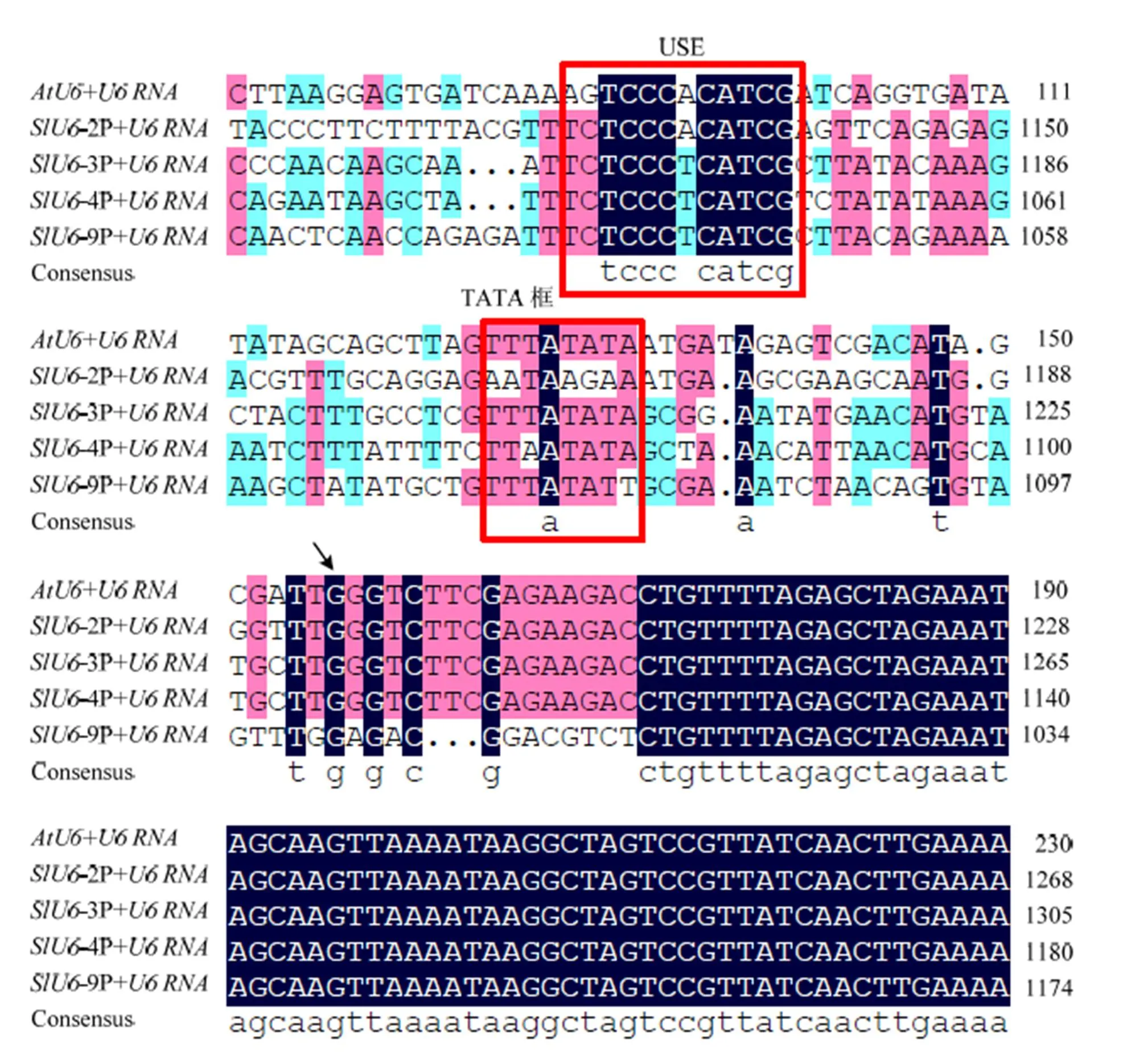
箭头为U6 RNA的转录起始位点 Arrow indicate the transcription start site of U6 RNA
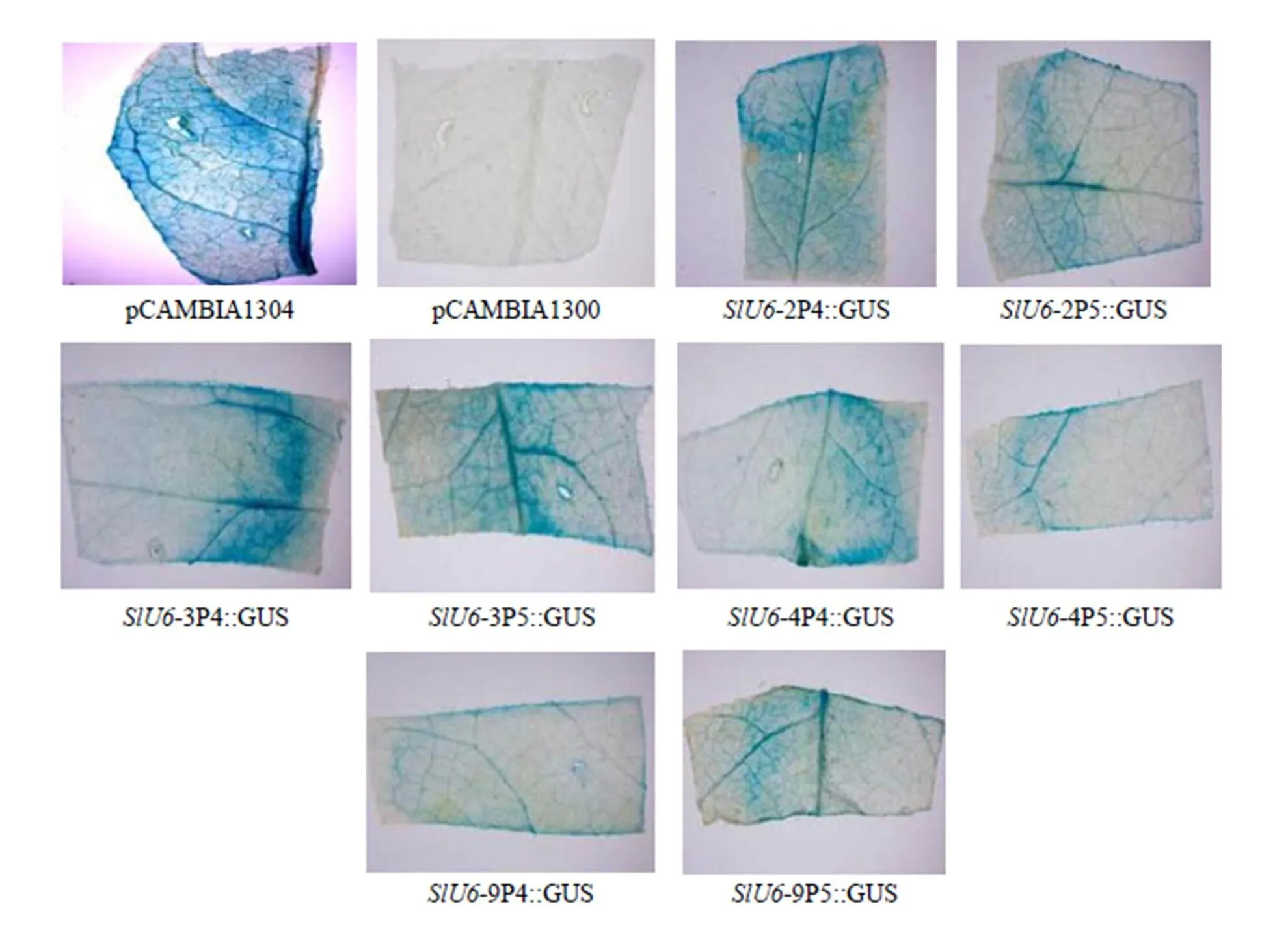
图6 不同截短SlU6启动子驱动GUS在番茄叶片中的瞬时表达
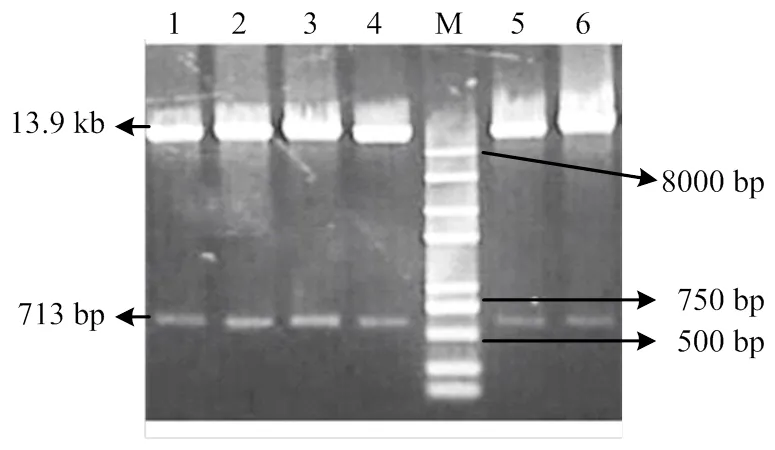
M:2 kb PlusⅡ标准分子量2 kb PlusⅡDNA Marker;1:构建SlU6-2p4- sgRNA::Cas9载体,以番茄EDR1设计3个为靶序列,1、2、3分别为带有Bgl II、MfeⅠ、NdeⅠ酶切位点;4、5、6以番茄MLO1设计3个靶序列分别含有Mfe I、Sap I、Pst I酶切位点;1: Construct SlU6-2p4-gRNA::Cas9 vector, which have three target sequences in EDR1respectively, lanes 1, 2, 3 contains BglⅡ, MfeⅠ, NdeⅠ enzyme respectively and another three target sequences in MLO1, respectively; lanes 4, 5, 6 contains MfeⅠ, SapⅠ, PstⅠenzyme sites, respectively
上述结果证明番茄的-2P4启动子可以使CRISPR/ Cas9基因编辑载体系统在番茄中实现对内源基因编辑的功能。
3 讨论
由RNA介导的CRISPR-Cas9基因编辑系统,作为一项全新的第三代人工核酸酶技术,其载体构建简单、成本消耗低,目前已成功地应用在拟南芥[16]、棉花[20]、玉米[21]、水稻[22]、烟草[23]、大豆[24]等物种的基因组定点编辑研究中[25],不论是锌指蛋白核酸酶或TALEN技术,还是近几年的研究热点CRISPR-Cas9基因编辑技术,其介导的基因定点突变的原理都是在基因组中定点产生DNA双链断裂,随后诱发机体启动DNA损伤修复机制从而造成DNA序列的改变。在CRISPR-Cas9系统中,RNA对应的启动子常被用于驱动sgRNA的转录,是CRISPR-Cas9系统中重要的元件。RNA由RNA聚合酶Ⅲ识别启动子来进行转录,其转录活性较高并且具有非常明确的转录起始位点[26-28]。利用植物内源的启动子,已在多种植物中建立了CRISPR-Cas9系统,如JACOBS等[29]运用大豆内源启动子成功修饰了大豆内源的9个基因,在大豆中建立了CRISPR-Cas9体系;XING[15]、LI[16]等运用拟南芥启动子成功实现了内源基因的编辑,在拟南芥中建立了CRISPR/ Cas9 基因编辑技术体系。虽然BROOKS等[30]在番茄中利用拟南芥启动子驱动sgRNA,成功实现了内多位点的编辑,建立了番茄的CRISPR/Cas9基因编辑体系。但利用番茄内源启动子的CRISPR/ Cas9系统是否也能实现内源基因的编辑,目前并不清楚。另外近两年研究表明利用多个或启动子驱动多个sgRNA同时靶定在同一条染色体上,可造成染色体大片段的缺失而引起成簇基因的删除。ZHANG等[31]用不同的启动子转录6个sgRNA,在拟南芥中同时敲除了ABA的受体PYLs家族中的多个成员;ZHOU等[32]在水稻中的研究结果表明,靶位点除了发生小片段的碱基突变外,也检测到染色体发生了115—245 kb不同长度的大片段缺失。因此鉴定更多有转录活性的启动子,对于CRISPR/Cas9系统的实际应用也具有重要价值。
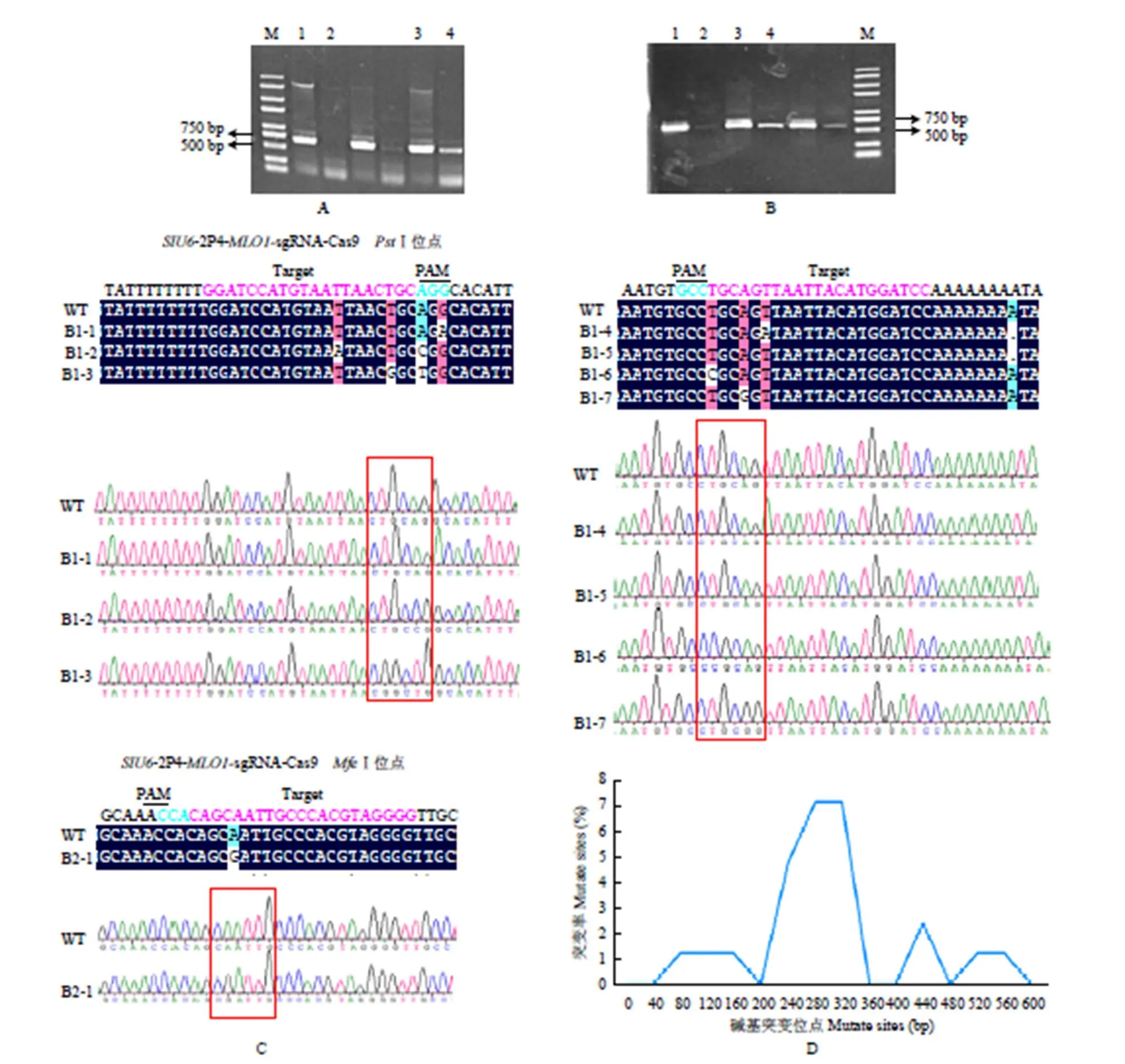
M:2 kb plusⅡDNA标准分子量2 kb plusII DNA Marker;A、B:采用RE/PCR的方法检测MLO靶位点突变体检测。空白对照为不带有sgRNA的CRISPR-Cas9基因编辑载体;A为MLO1内MfeⅠ靶位点突变的检测。1和2分别为空白对照转化原生质体后DNA直接PCR和MfeⅠ酶切后PCR产物;3和4分别为:带有MLO1-MfeⅠsgRNA的CRISPR-Cas9系统转化原生质体后DNA直接PCR和MfeⅠ酶切后PCR产物;B为MLO1基因内PstⅠ靶位点突变的检测。1和2分别为空白对照转化原生质体后DNA直接PCR和PstⅠ酶切后PCR产物;3和4分别为:带有MLO1-PstⅠsgRNA的CRISPR-Cas9系统转化原生质体后DNA直接PCR和PstⅠ酶切后PCR产物;C:番茄MLO1内两个不同靶位点(PstⅠ和MfeⅠ)测序的结果。上图是靶位点序列比对;下图是靶位点序列测序峰图,红色方框为内切酶识别位点。D:MLO1内MfeⅠ靶位点突变频率分布结果图,红色方框为含靶序列的突变位点A, B: Detection of mutation in MLO1 by RE/PCR assays; The negative control is CRISPR-Cas9 system without sgRNA. A: Detection of mutation in MfeⅠtarget site of MLO1 gene. Lanes 1 and 2 represent PCR products from MfeⅠ-undigested and digested negative control genomic DNA, respectively; Lanes 3 and 4 represent respectively PCR products from MfeⅠ-undigested and digested sample genomic DNA that was prepared from protoplast transformed by CRISPR-Cas9 system with MLO1-MfeⅠsgRNA; B: Detection of mutation in PstⅠtarget site of MLO1 gene. Lanes 1 and 2 represent PCR products from PstⅠ-undigested and digested negative control genomic DNA, respectively; Lanes 3 and 4 represent respectively PCR products from PstⅠ-undigested and digested sample genomic DNA that was prepared from protoplast transformed by CRISPR-Cas9 system with MLO1- PstⅠsgRNA; C: Sequencing of two target sites (PstⅠand MfeⅠ) in tomato MLO. Sequence alignment of target sites is above the picture; Sequencing peaks figure of target site is below. The red box indicates that the region recognized by restriction enzyme; D: Mutation efficiency of MLO1 in MfeⅠtarget site, respectively. The red squared indicates mutation sites contains target sequence
本研究从‘中蔬四号’番茄中克隆的4个番茄启动子启动区都含有两个序列保守的启动元件TATA框和USE元件,并且这两个元件之间的距离也比较固定,虽然间隔区的序列并不重要,但其距离大小对帮助几何对称的RNA聚合酶结合到一定位点是很重要的。依据启动元件的位置,本研究分别对4个启动子进行两种长度的截短,共得到8个不同长度的启动子,与报告基因融合后进行检测,结果表明这4种番茄启动子均具有转录活性。当这4种启动子截短到200 bp左右时,依然能驱动报告基因的表达。这显示出番茄的启动子相对较短时也具有转录活性,与在其他作物中克隆的启动子特点相同[33]。棉花的启动子截短为105 bp时,其转录活性并没有降低[34]。NEKRASOV等[35]在基因组编辑技术研究中甚至使用了拟南芥79 bp长度的启动子,并且成功实现了基因编辑。这些结果暗示了番茄的启动子可以克隆的更短。但是否可以仅保留启动子-60 bp位置的USE元件和-30 bp位置的TATA框还能具有转录活性,这需要进一步研究。
虽然已报道许多不同物种的启动子在CRISPR-Cas9系统中成功得到了应用,但同样的启动在亲缘关系较远物种中不一定适用,并且目前适用于番茄CRISPR-Cas9系统的内源启动子并无报道,筛选出在番茄中有功能活性的启动子很有必要。本研究结果表明番茄内源-2P4启动子实现了CRISPR-Cas9介导的番茄基因组靶向编辑。克隆测序结果发现靶位点的突变多为碱基的替换,这与棉花原生质体中的基因编辑结果类似。CHEN等[5]设计了两个sgRNA,在棉花原生质体中成功实现了对和位点内的靶向修饰,其结果在原生质体中也多为碱基的替换,而在稳定转化的棉花植株中却出现较多的碱基缺失和插入。原因可能与原生质体自身的细胞状态和所处的环境有关,这与活体组织中的细胞所处的状态和环境是不同的,可能在原生质体细胞中同源修复机制的活性被大大激活。另一个可能的原因是原生质体中Cas9和sgRNA是瞬时表达,靶位点产生的碱基替换突变,由于没有足够多的Cas9和sgRNA而不能继续被编辑修饰。而在稳定转化的植株中Cas9和sgRNA可以持续表达,靶位点产生的碱基替换突变可以被继续编辑,直到产生的突变不能被sgRNA有效地识别而终止。在研究中同时也构建了以为靶序列的CRISPR/Cas9基因编辑载体,但瞬时转化番茄原生质体后并没有检测到靶基因被编辑的现象,这与在线虫中的一些研究结果相同[36],目前原因并不清楚。单碱基的改变是否是因为PCR扩增过程中碱基随机配对错误产生的突变?为排除这个可能性,对样品基因组DNA酶切前扩增的PCR片段序列进行了克隆,经测序分析,结果显示靶位点碱基的突变频率要远远高于其两侧其他位点的突变频率,表明靶序列区碱基的突变确实是CRISPR/Cas9系统靶向编辑的结果。
4 结论
从‘中蔬四号’番茄品种中克隆了4种8个不同长度的启动子,其长度分别为452、202、448、206、433、190、448和218 bp,这8个启动子在番茄叶片中都具有转录活性。其中以-2P4为启动子驱动sgRNA,以白粉病相关基因为靶序列的CRISPR/Cas9基因编辑载体成功地实现对番茄内源基因的编辑。
[1] Sato S, Tabata S, Hirakawa H, Asamizu E, Shirasawa K, Isobe S, Kaneko T, Egholm M. The tomato genome sequence provides insights into fleshy fruit evolution., 2012, 485(7400): 635-641.
[2] Hisamatsu S, Sakaue M, Takizawa A, Kato T, Kamoshita M, Ito J, Kashiwazaki N.Knockout of targeted gene in porcine somatic cells using zinc-finger nuclease., 2015, 86(2): 132-137.
[3] Sun N, Zhao H. Transcription activator-like effector nucleases (TALENs): A highly efficient and versatile tool for genome editing., 2013, 110(7): 1811-1821.
[4] Chen K, Gao C. Targeted genome modification technologies and their applications in crop improvements., 2014, 33(4): 575-583.
[5] Chen X, Lu X, Shu N, Wang S, Wang J, Wang D, Guo L, Ye W. Targeted mutagenesis in cotton (L.) using the CRISPR/Cas9 system., 2017, 7: 44304.
[6] Li C, Unver T, Zhang B. A high-efficiency CRISPR/Cas9 system for targeted mutagenesis In Cotton (L.)., 2017, 7: 43902.
[7] Liu X, Xie C, Si H, Yang J. CRISPR/Cas9-mediated genome editing in plants.,2017, 3: 9.
[8] Friedland A E, Tzur Y B, Esvelt K M, Colaiácovo M P, Church G M, Calarco J A. Heritable genome editing in C. elegans via a CRISPR-Cas9 system., 2013, 10(8): 741-743.
[9] Wang M B, Helliwell C A, Wu L M, Waterhouse P M, Peacock W J, Dennis E S.Hairpin RNAs derived from RNA polymerase II and polymerase III promoter-directed transgenes are processed differently in plants., 2008, 14(5): 903-913.
[10] Domitrovich A M, Kunkel G R. Multiple, dispersed human U6 small nuclear RNA genes with varied transcriptional efficiencies., 2003, 31(9): 2344-2352.
[11] Jia H, Wang N. Targeted genome editing of sweet orange using Cas9/sgRNA., 2014, 9(4): e93806.
[12] 雷建峰, 伍娟, 陈晓俊, 於添平, 倪志勇, 李月, 张巨松, 刘晓东.棉花花粉中高效转录U6启动子的克隆及功能分析. 中国农业科学, 2015, 48(19): 3794-3802.
Lei J F, Wu J, Chen X J, Yu T P, Ni Z Y, Li Y, Zhang J S, Liu X D. Cloning and functional analysis of cotton U6 promoter with high transcription activity in cotton pollen.2015, 48(19):3794-3802. (in Chinese)
[13] Simmen K A, Mattaj I W. Complex requirements for RNA polymerase III transcription of the Xenopus U6 promoter., 1990, 18(19): 5649-5657.
[14] 刘玟杉. 基于CRISPR/Cas9的拟南芥多基因编辑及其在基因功能研究中的应用[D]重庆: 重庆大学, 2015.
Liu W S. CRISPR-Cas9-mediated multiplex genome editing inand the application in gene functions study [D]. Chongqing: Chongqing University, 2015. (in Chinese)
[15] Xing H L, Dong L, Wang Z P, Zhang H Y, Han C Y, Liu B, Wang X C, Chen Q J. A CRISPR/Cas9 toolkit for multiplex genome editing in plants., 2014, 14(1): 327.
[16] Li J F, Norville J E, Aach J, McCormack M, Zhang D, Bush J, Church G M, Sheen J. Multiplex and homologous recombination-mediated genome editing inandusing guide RNA and Cas9., 2013, 31(8): 688-691.
[17] Erijman A, Shifman J M, Peleg Y. A single-tube assembly of DNA using the transfer-PCR (TPCR) platform., 2014, 1116: 89-101.
[18] Yoo S D, Cho Y H, Sheen J.mesophyll protoplasts: a versatile cell system for transient gene expression analysis., 2007, 2(7): 1565-1572.
[19] Lu Y M, Chen X, Wu Y X, Wang Y P, He Y Q, Wu Y. Directly transforming PCR-amplified DNA fragments into plant cells is a versatile system that facilitates the transient expression assay., 2013, 8(2): e57171.
[20] Janga M R, Campbell L M, Rathore K S. CRISPR/Cas9- mediated targeted mutagenesis in upland cotton (L.)., 2017, 94: 349-360.
[21] Liang Z, Zhang K, Chen K, Gao C. Targeted mutagenesis inusing TALENs and the CRISPR/Cas system., 2014, 41(2): 63-68.
[22] Shan Q, Wang Y, Li J, Gao C. Genome editing in rice and wheat using the CRISPR/Cas system., 2014, 9(10): 2395-2410.
[23] Jiang W, Zhou H, Bi H, Fromm M, Yang B. Demonstration of CRISPR/Cas9/sgRNA-mediated targeted gene modification in,tobacco,sorghum and rice., 2013, 41(20): e188.
[24] Jacobs T B,LaFayette P R,Schmitz R J,Parrott W A. Targeted genome modifications in soybean with CRISPR/Cas9., 2015, 15(1): 16.
[25] Cao H X, Wang W, Le H T T, Vu G T H. The Power of CRISPR-Cas9-Induced Genome Editing to Speed Up Plant Breeding., 2016.
[26] Cong L, Ran F A, Cox D, Lin S, Barretto R, Habi b, Zhang F. Multiplex genome engineering using CRISPR/Cas systems., 2013, 339(6121): 819-823.
[27] Mali P, Yang L, Esvelt K M, Aach J, Guell M, DiCarlo J E, Church G M. RNA-guided human genome engineering via Cas9., 2013, 339(6121): 823-826.
[28] Belhaj K, Chaparro-Garcia A, Kamoun S, Nekrasov V. Plant genome editing made easy: Targeted mutagenesis in model and crop plants using the CRISPR/Cas system., 2013, 9(1): 39.
[29] Jacobs T B, LaFayette P R, Schmitz R J, Parrott W A. Targeted genome modifications in soybean with CRISPR/Cas9., 2015, 15(1): 16.
[30] Brooks C, Nekrasov V, Lippman Z B, Van Eck J. Efficient gene editing in tomato in the first generation using the clustered regularly interspaced short palindromic repeats/CRISPR-associated9 System ., 2014, 166(3): 1292-1297.
[31] Zhang Z, Mao Y, Ha S, Liu W, Botella J R, Zhu J K. A multiplex CRISPR/Cas9 platform for fast and efficient editing of multiple genes in, 2016, 35(7): 1519-1533.
[32] Zhou H, Liu B, Weeks D P, Spalding M H, Yang B. Large chromosomal deletions and heritable small genetic changes induced by CRISPR/Cas9 in rice., 2014, 42(17): 10903.
[33] Gasiunas G, Barrangou R, Horvath P, Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria., 2012, 109(39): 2579-2586.
[34] 雷建峰, 李月, 徐新霞, 阿尔祖古丽·塔什, 蒲艳, 张巨松, 刘晓东. 棉花不同启动子截短克隆及功能鉴定. 作物学报, 2016, 42(5): 675-683.
Lei J F, Li Y, Xu X X, Tashi A, Pu Y, Zhang J S, Liu X D. Cloning and functional analysis of different truncated GbU6 promoters in cotton.,2016, 42(5): 675-683.(in Chinese)
[35] Belhaj K, Chaparro-Garcia A, Kamoun S, Nekrasov V. Plant genome editing made easy: Targeted mutagenesis in model and crop plants using the CRISPR/Cas system., 2013, 9(1): 39.
[36] Farboud B, Meyer B J. Dramatic enhancement of genome editing by CRISPR/Cas9 through improved guide RNA design., 2015, 199(4): 959-971.
(责任编辑 赵伶俐)
DifferentPromoters Cloning and Establishment of CRISPR/Cas9 Mediated Gene Editing System in Tomato
PU Yan, LIU Chao, LI Ji-Yang, AERZU GULI·TaShi, HU Yan, LIU XiaoDong
(College of Agronomy, Xinjiang Agricultural University/Laboratory of Agricultural Biotechnology of Xinjiang Agricultural University, Urumqi 830052)
【Objective】promoter is a vital element for the transcription of sgRNA inthe CRISPR/Cas9 system. It is necessary to clone some endogenouspromoters with high transcription activity and construct CRISPR/Cas9 vector, which could provide a strong technical basis for functional genomics and molecular breeding in tomato. 【Method】 Four different tomatopromoters were cloned by first round of PCR amplification from tomato cultivar Zhongshu 4. Each U6 promoter with two different length was truncated and used to construct plant expression vector carried SlU6 promoter, respectively. The Eightfusion expression vectors were transformed into tomato leaves by agroinfiltration. According to the degree ofstaining, the promoter with high transcription activity was selected to construct the CRISPR/Cas9 gene editing vector with target sequence from powdery mildew-related geneandThese gene editing vectors were transformed into tomato protoplast by PEG method. The mutation of endogenous target genes in each transformed tomato protoplast was analyzed by a restriction enzyme PCR (RE-PCR) assay. Finally, the types of endogenous gene mutation were analyzed by sequencing. The efficiency of the CRISPR/Cas9 system based on tomato endogenous U6 promoter was verified by the frequency distribution map of mutant loci. 【Result】4 kinds and 8 different lengths of tomatopromoters were obtained by two rounds of PCR. Their length was 452, 202, 448, 206, 433, 190, 448 and 218 bp, respectively. After sequences analysis, results showed that the four tomatopromoters also contained the USE motif and TATA box which were found inpromoters. The construction offusion expression vectors driven by corresponding truncated tomatopromoters were done and transformed into tomato leaves. Thehistochemical staining showed that the transformed tomato leaves were dyed blue, which indicated that all 8promoters have transcription activity. The6-2P4 promoter was chose to drive sgRNA transcription and construct the CRISPR/Cas9 system with target sequence fromandrespectively. The result showed that endogenous6-2P4 promoter could drive sgRNA transcription and genewas edited successfully in tomato. Sequence analysis revealed that all types of gene mutations are base substitution and the hotspot of mutation only exists in the target region of endogenous gene. 【Conclusion】4 kinds of6 promoters with high transcription efficiency were obtained from tomato. The established CRISPR/Cas9 system based on SlU6-2 promoter could successfully achieve the editing of endogenous genes in tomato.
CRISPR/Cas9;6 promoter; clone; tomato; gene editing
2017-06-21;
2017-07-08
国家自然科学基金(31560534)
蒲艳,E-mail:603314289@qq.com。
刘晓东,Tel:18290812916;E-mail:xiaodongliu75@aliyun.com
猜你喜欢
环球时报(2022-09-20)2022-09-20
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
江西农业学报(2021年4期)2021-04-20
兽医导刊(2020年13期)2020-12-31
今日农业(2020年24期)2020-12-15
三农资讯半月报(2020年11期)2020-06-21
蔬菜(2019年5期)2019-01-04
小资CHIC!ELEGANCE(2015年14期)2015-09-23
小资CHIC!ELEGANCE(2015年15期)2015-09-01
