
UHPLC-MS/MS测定罗非鱼中硝基呋喃类代谢物残留量的不确定度评定
2018-01-10 08:45林功师
中国渔业质量与标准 2017年6期
林功师
(厦门市海洋与渔业研究所, 福建 厦门 361008)
UHPLC-MS/MS测定罗非鱼中硝基呋喃类代谢物残留量的不确定度评定
林功师
(厦门市海洋与渔业研究所, 福建 厦门361008)
为判断罗非鱼中硝基呋喃类代谢物残留量测定中不确定度的主要来源,本研究参照JJF1059.1—2012《测量不确定度的评定与表示》等相关理论,采用农业部783号公告-1-2006的前处理方法进行样品处理,并根据公告中的结果计算公式建立相应的数学模型,对罗非鱼中硝基呋喃类代谢物残留量的测定结果中产生的各不确定度来源进行分析和计算。当硝基呋喃类代谢物的添加水平为1.00μg/kg时,4种硝基呋喃类代谢物结果可表示为:呋喃西林代谢物CSEM为(0.989±0.168)μg/kg,k=2;呋喃妥因代谢物CAHD为(1.009±0.165)μg/kg,k=2;呋喃它酮代谢物CAMOZ为(0.991±0.169)μg/kg,k=2;呋喃唑酮代谢物CAOZ为(0.986±0.180)μg/kg,k=2。研究表明,在确定的实验条件下,本方法的不确定度主要来自于标准溶液和内标溶液配制、标准曲线拟合和加标回收率。研究结果对实际操作中减少不确定度,提高检测结果的准确性和可靠性具有重要意义。[中国渔业质量与标准,2017,7(6):48-57]
超高效液相色谱-串联质谱法(UHPLC-MS/MS);罗非鱼;硝基呋喃类代谢物;不确定度
硝基呋喃类药物是人工合成的含有硝基的一类广谱抗菌性药物,因其具有价格低廉、杀菌能力强等优点,曾被应用于水生动物养殖环境杀菌消毒和家畜、水产等养殖过程中动物疾病的预防与治疗[1-2]。但该类药物在生物体内代谢很快,其代谢产物能与机体内的蛋白质稳定的结合,并诱发机体发生基因突变造成,致癌、致畸等危害[3-5]。因此,世界许多国家对硝基呋喃类药物的控制变得很严格。1997年欧盟将硝基呋喃类药物列为A类违禁药物,随后美国、日本等国家也制定了相应的法规[6]。2003年欧盟发布2003/181/EC条例,要求检测硝基呋喃类药物及其代谢物残留量的各种检测方法最低检测限需达到1.0μg/kg[7]。中国农业部也于2002年12月发布第235号公告及2005年10月发布第560号公告[8-9],将硝基呋喃类药物列为禁止使用的兽药,并要求在动物性食品中不得检出。2010年3月卫生部也明确将硝基呋喃类药物(主要成分:呋喃西林、呋喃它酮、呋喃唑酮、呋喃妥因)列入食品中可能违法添加的非食用物质名单中[10]。目前检测水产品中硝基呋喃类代谢物的方法主要有免疫胶体金法[11]、酶联免疫法[12]、高效液相色谱法[13]和液相色谱串联质谱法[14-15]。其中,免疫胶体金法主要用于水产品中硝基呋喃类代谢物的快速检测,它只能做定性判定,无法做定量检测;酶联免疫法和高效液相色谱法虽可做定量检测,但出现假阳性的概率比较大。液相色谱串联质谱法是目前国际上用于硝基呋喃类代谢物确证的通用方法。
测量不确定度是一种表示被测量值离散程度的重要参数,是评价测量结果和测量水平的参数指标[16]。检测和校准实验室能力的通用要求(GB/T27025—2008)中明确规定应具有评定测量不确定度的程序[17]。近年来,人们对检测数据的准确性和可靠性要求越来越高,实验室计量认证评审等体现实验室检测能力的资质认证过程对数据的要求也在提高,因此分析检测过程中数据的不确定度成为了检测过程中必须完成的一项工作。根据《化学分析中不确定度的评估指南》[18]和《测量不确定度评定与表示》[19]等技术规范,常晨阳等[20]和陈国花[21]对动物源性食品中硝基呋喃类代谢物中的一种目标物进行了不确定度评定;何太喜等[22]采用GB/T21311—2007的检测方法[23]对鳗鱼肉中硝基呋喃类代谢物进行不确定度评定;祝子铜等[15]、邢丽红等[24]、郭丽娜等[25]根据农业部783号公告-1-2006[26]采用液相色谱串联质谱法对水产品中硝基呋喃类代谢物的不确定度进行评定;李绪鹏等[14]根据农业部783号公告-1-2006[24]采用超高效液相色谱串联质谱法测定水产品中硝基呋喃类代谢物的不确定度,但未充分考虑温度因素对不确定度评定的影响。为更全面地考察本实验室在检测硝基呋喃类代谢物的过程中存在的可能影响检测结果的不确定因素,本研究采用农业部783号公告-1-2006的检测方法,选用近年来农业部农产品质量安全例行监测(风险监测)中均要求监测的水产品品种之一——罗非鱼作为研究对象,对本实验室检测罗非鱼中4种硝基呋喃代谢物残留量的过程中可能影响检测结果的不确定度进行评定,分析不确定度的主要来源,以保证检测结果的准确性和可靠性,从而提高检测数据的质量。
1 材料和设备
1.1 仪器与设备
超高效液相色谱(1290 Infinity UHPLC,美国 Agilent 公司);三重四级杆串联质谱仪(AB SciexTriple Quad 4500,美国AB公司,配有电喷雾 ESI 离子源);DKZ-2往复式水浴恒温振荡器(上海一恒科学仪器有限公司);MS3 Digital 漩涡振荡器(德国IKA公司);BS110S电子天平和TE212电子天平(德国Sartorius公司);MG-2100氮吹仪(日本东京理化器械株式会社);Sorvall Stratos高速冷冻离心机(美国Thermo公司);Milli-Q Gradient超纯水仪(美国Millipore公司);Multi 9310台式pH分析仪(德国WTW公司);20、 25、 100、 1 000 μL移液器(法国Gilson公司)。
1.2 试剂与材料
呋喃西林代谢物盐酸盐SEM·HCl纯度 99.5%,德国Dr公司;呋喃妥因代谢物盐酸盐AHD·HCl纯度 99.7%,德国Dr Eppendorf公司;呋喃唑酮代谢物AOZ纯度 99.7%,Sigma 公司;呋喃它酮代谢物AMOZ 纯度 99.4%,Sigma 公司;呋喃西林代谢物同位素标准品SEM-13C-15N2纯度98.4%,Sigma 公司;呋喃妥因代谢物同位素标准品AHD-13C3纯度 99.3%,Sigma 公司;呋喃唑酮代谢物氘代物AOZ-D4纯度 99.0%,Sigma 公司;呋喃它酮代谢物氘代物AMOZ-D5纯度 99.5%,Sigma 公司;甲醇、乙酸乙酯、二甲亚砜、 2- 硝基苯甲醛,甲酸为色谱纯,购自 Fisher Scientific 公司;盐酸为优级纯、磷酸氢二钾和乙酸铵为分析纯,购自上海国药集团;实验用水为超纯水。
实验用的水产品样品为采购于水产批发市场的罗非鱼(Oreochromismossambicus),采样规格为每尾体重约1.0 kg,采样量为3尾。
1.3 方法
1.3.1 标准溶液的配置
标准储备溶液:分别准确称取AOZ 9.9 mg、SEM·HCL 15.4 mg、AMOZ 10.2 mg、AHD·HCL 13.1 mg、AOZ-D49.9 mg、SEM-13C-15N29.9 mg、AMOZ-D510.1 mg、AHD-13C310.1 mg,用甲醇溶解并定容至10 mL棕色容量瓶中,配制成1.0 mg/mL标准储备溶液。
混合标准储备溶液:用1 000 μL移液器各移取1.0mL 1.0 mg/mL标准储备液于10 mL容量瓶中,用超纯水定容至刻度,得到100.0 μg/mL的混合标准储备液。采用同样的方法依次十倍稀释,分别制得10.0、 1.0 μg/mL混合标准储备液和100.0、 10.0 μg/L混合标准工作液。
混合内标工作溶液:用1 000 μL移液器各移取1.0 mL 1.0 mg/mL内标标准储备液于10 mL容量瓶中,用超纯水定容至刻度,得到100 μg/mL的混合内标储备液。采用同样的方法依次十倍稀释,分别制得10.0、 1.0 μg/mL的混合内标储备液和100.0 μg/L的混合内标工作液。
样品加标上机液:取2.00 g空白样品至 50 mL离心管,分别加入20 μL 100.0 μg/L混合标准工作液和50 μL 100.0 μg/L混合内标工作溶液,按1.3.2 步骤操作,制成样品加标上机液。
标准曲线工作液:分别准确吸取 10.0 μg/L混合标准工作液10、 25、 50、 100 μL和准确吸取 100.0 μg/L混合标准工作液25、 50、 100 μL至7个 50 mL离心管中,分别加入50 μL 100.0 μg/L混合内标工作溶液,除不加样品外其余步骤按1.3.2样品前处理方法操作,制成0.10、 0.25、 0.50、 1.0、 2.5、 5.0、 10.0 μg/L的标准曲线工作液,待测。
1.3.2 样品前处理方法
称取样品2.00 g于50 mL离心管中,加入100.0 μg/L混合内标工作溶液50 μL,充分混合50 s,再加入0.1 mol/L盐酸溶液5.0 mL 和0.05 mol/L 2-硝基苯甲醛溶液150 μL,涡旋振荡50 s后,置于恒温水浴振动器中37 ℃避光振荡16 h,进行充分的衍生化反应。取出离心管加入适量1.0 mol/L磷酸氢二钾溶液,使离心管中的溶液pH为7.0~7.5。再加入乙酸乙酯4.0 mL,涡旋振荡50 s,13 000 r/min离心3 min,将上清液转移至新的15 mL离心管中;再加入乙酸乙酯4.0 mL重复提取目标物一次,合并上清液,40 ℃氮气吹干。准确加入5%甲醇溶液1.00 mL,溶解残留物,4 500 r/min离心3 min,取清液,过0.22 μm滤膜,待测。
1.3.3 液相色谱-串联质谱条件
色谱柱为Zorbax Eclipse Plus-C18(2.1 mm×100 mm, 1.8 μm);流动相A为甲醇,流动相B为5 mmol/L乙酸铵;柱温40 ℃;流速0.25 mL/min;进样量20 μL;液相色谱流速及梯度洗脱程序见表 1。离子源为电喷雾离子源 ESI,毛细管电压为5 500V,离子源气流量为55.0 mL/min,锥孔气流量35.0 mL/min。采用正离子扫描方式,多反应监测模式(MRM) 。质谱定性定量离子对信息见表 2。

表1 液相色谱梯度洗脱条件Tab.1 LC gradient elution conditions
注:A为甲醇,B为5 mmol·L-1乙酸铵。

表2 目标化合物液质测定条件Tab.2 Mass spectrometric parameters for target compounds
2 结果与分析
2.1 不确定度的数学模型
根据农业部783号公告-1-2006的测定方法及结果计算公式,建立检测中硝基呋喃类代谢物含量测定的数学模型,如式(1)所示。

式(1)
式(1)中:X为检测样品中硝基呋喃代谢物的含量(μg/kg);Ax为提取的样品待测液中的目标物与同位素内标峰面积比值;Cs为标准溶液中硝基呋喃代谢物的质量浓度(μg/L);As为标准溶液中的目标物与同位素内标峰面积比值;V为提取的样品待测液最终定容体积(mL);m为检测样品的质量(g),R为回收率。
合成相对标准不确定度计算的计算公式见式(2)。
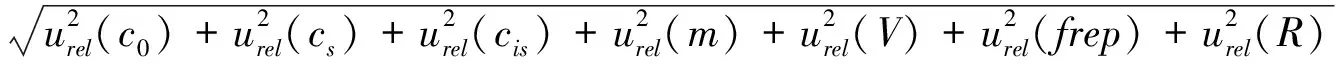
式(2)
式(2)中:urel(c)为合成相对标准不确定度,urel(c0)为标准曲线拟合的相对标准不确定度,urel(cs)为标准溶液配制的相对标准不确定度,urel(cis)为同位素内标的相对标准不确定度,urel(m)为样品称量的相对标准不确定度,urel(V)为提取液浓缩后的最终定容体积的相对标准不确定度,urel(frep)为重复测量的相对标准不确定度,urel(R)为加标回收率的相对标准不确定度。
2.2 不确定度的来源分析及计算
2.2.1 标准曲线拟合引入的相对标准不确定度urel(c0)
本实验对4种硝基呋喃代谢物,制备了7个不同浓度点的标准工作液,每个浓度点检测 1 次,以外标峰面积/内标峰面积的值(Ai)为纵坐标,标准溶液浓度(Ci)为横坐标采用最小二乘法拟合,得出目标物的回归方程(y=a+bx)。由表3可以得出,AOZ的线性方程为y=0.327 53x+0.008 23(r=0.999 6),AMOZ的线性方程为y=0.274 13x+0.001 25(r=0.999 8),SEM的线性方程为y=0.284 51x+0.004 28(r=0.999 9),AHD的线性方程为y=0.196 97x+0.001 04(r=0.999 7)。
标准曲线拟合引入的不确定度见式(3)。

式(3)
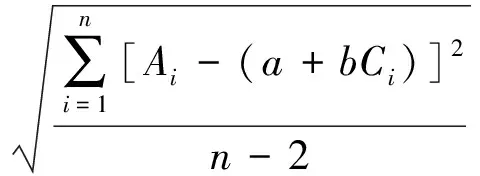
标准曲线拟合引入的相对标准不确定度urel(c0)见式(4)。u和urec(C0)计算结果见表4。
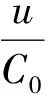
式(4)

表3 标准曲线测定中的相关系数Tab.3 Correlation coefficients of the linear curve

表4 相对不确定度Tab.4 Results of relative standard uncertainty
2.2.2 标准溶液配制过程引入的相对标准不确定度urel(cs)
标准溶液配制过程引入的相对标准不确定度的数学模型见式(5)。

式(5)
2.2.2.1 标准物质纯度引入的相对标准不确定度u21rel
AOZ标准品纯度为 99.7%,最大允差为±0.3%,其引入的相对标准不确定度为:
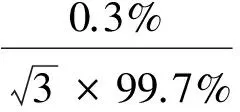
AMOZ标准品纯度为99.4%,最大允差为±0.3%,其引入的相对标准不确定度为:
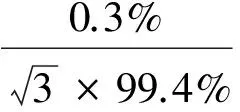
SEM标准品纯度为99.5%,最大允差为±1.0%,其引入的相对标准不确定度为:
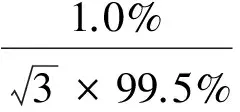
AHD标准品纯度为99.7%,最大允差为±1.0%,其引入的相对标准不确定度为:

2.2.2.2 标准物质称量引入的相对标准不确定度u22rel


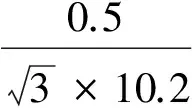
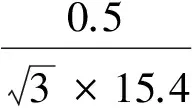

2.2.2.3 标准储备液配制中引入的相对标准不确定度u23rel
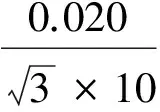
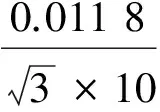

2.2.2.4 混合标准储备液配制中引入的相对标准不确定度u24rel



从1.00 mg/mL逐级稀释到10.0 μg/L的过程中共使用了5次1 000 μL移液器和5次10 mL A 级单标容量瓶,故其引入的相对标准不确定度为:

。
2.2.2.5 标准曲线工作液配制中引入的相对标准不确定度u25rel
本研究中标准曲线点共有7个,在配制过程中所使用的量器为20、25、100 μL移液器,其中20 μL移液器使用1次,25 μL移液器使用2次、100 μL移液器使用4次。


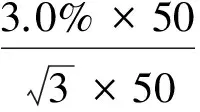

因此,标准曲线工作液配制中引入的相对标准不确定度为:
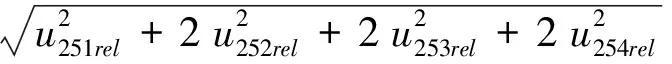
=0.063 7
根据以上计算的各分量及公式(5)可以计算出标准溶液配制过程中的相对标准不确定度urel(cs):urel(csAOZ)=0.071 3,urel(csAMOZ)=0.071 0,urel(csSEM)=0.067 9,urel(csAHD)=0.068 9。
2.2.3 混合内标工作溶液配制中引入的相对标准不确定度urel(cis)
混合内标工作溶液配制中引入的相对标准不确定度的数学模型见式(6)。
式(6)
同位素内标(AOZ-D4)纯度为(99.0±0.3)%,AMOZ-D5纯度为(99.5±0.3)%,SEM-13C-15N2纯度为(98.4±0.5)%,AHD-13C3纯度为(99.3±0.3)%,纯度的相对标准不确定度为u31rel。其天平称量(u32rel)、溶液稀释(u33rel)和定容(u34rel)等一系列溶液配制过程引入的相对标准不确定度计算方法与标准溶液2.2.2.1~2.2.2.4同理,其结果依次分别是:


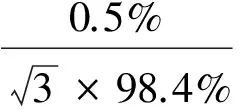
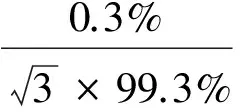
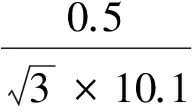
u33rel=u23rel=0.001 33。
混合内标工作溶液从1.00 mg/mL逐级稀释到100.0 μg/L的过程中共使用了4次1 000 μL移液器和4次10 mL A 级单标容量瓶,故其引入的相对标准不确定度为:

将同位素内标各项分量代入公式 (6),计算结果如下:urel(cis(AOZ))=0.038 4,urel(cis(AMOZ))=0.037 9,urel(cis(SEM))=0.038 4,urel(cis(AHD))=0.037 9。
2.2.4 样品称量过程中引入的相对标准不确定度urel(m)

2.2.5 提取液浓缩后的最终定容体积引入的相对标准不确定度urel(V)

=0.000 148。
提取液定容引入的相对标准不确定度为:
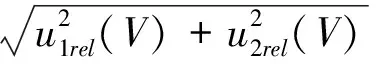
2.2.6 重复测量引入的相对标准不确定度urel(frep)
在同一未检出样品中添加同一浓度的混合标准溶液,按照1.3方法进行处理和上机测定,重复测定 6次,用贝塞尔公式(式7)进行计算,结果见表5。

式(7)

式(8)
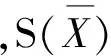

表5 重复测定结果和重复测定引入的相对标准不确定度Tab.5 Results of repeated determination and relative standard uncertainty
2.2.7加标回收率引入的相对标准不确定度urel(R)

式(9)

2.2.8 合成相对标准不确定度urel(c)及不确定度uc(X)
以上各项不确定度分量相互独立,不考虑分量间的相关性,将上述各值代入公式(2)中,则合成相对标准不确定度urel(c)和不确定度uc(X)最终检测结果取包含因子结果见表7。
uc(X)=urel(c)·X
式(10)
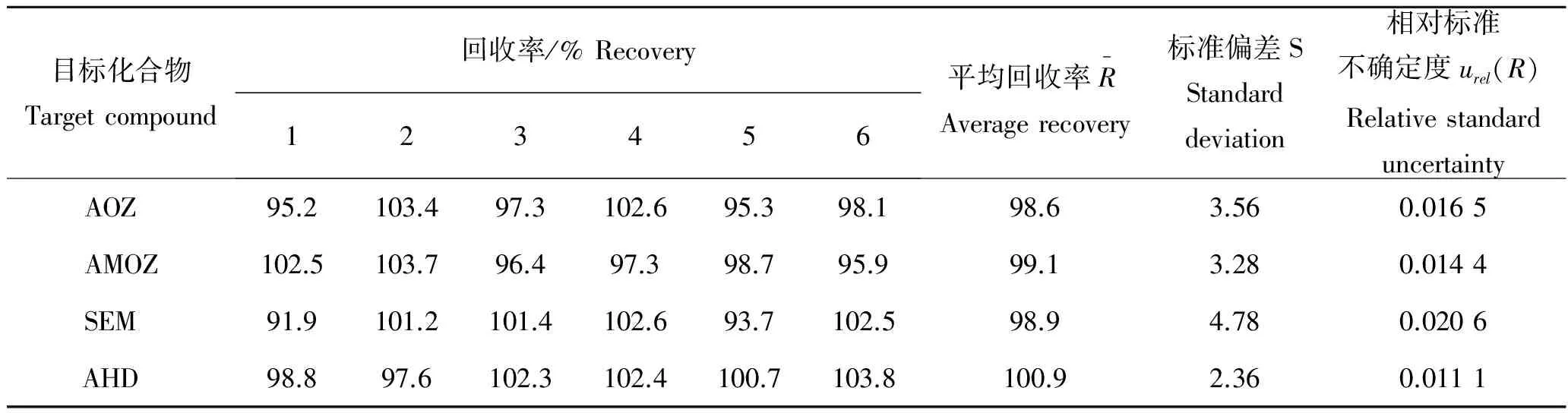
表6 加标回收率结果和加标实验引入的相对标准不确定度Tab.6 Results of the spiked recovery and relative standard uncertainty

表7 合成相对标准不确定度和不确定度Tab.7 Results of combined standard uncertainty and uncertainty
取包含因子k=2(近似95%置信概率),扩展不确定度U=kuc(X),则最终检测结果为:呋喃唑酮代谢物CAOZ=0.986 μg/kg,U=0.180 μg/kg,k=2;呋喃它酮代谢物CAMOZ=0.991 μg/kg,U=0.169 μg/kg,k=2;呋喃西林代谢物CSEM=0.989 μg/kg,U=0.168 μg/kg,k=2;呋喃妥因代谢物CAHD=1.009 μg/kg,U=0.165μg/kg,k=2。
3 结论
根据测量不确定度的评定方法,本研究对本实验室采用农业部 783 号公告-1-2006中液质联用法测定罗非鱼中硝基呋喃类代谢物残留量的不确定度来源进行分析,通过建立模型,评定和计算测量硝基呋喃类代谢物残留量过程中的各不确定度分量。结果表明,水产品中硝基呋喃代谢物残留量测定的不确定度主要来自于样品前处理过程的标准溶液和内标溶液的配制及使用、标准曲线拟合、加标回收率,其次为测量重复性和样品称量。因此,在日常检测水产品中硝基呋喃类代谢物残留量的过程中,应尽量选用精度较高的玻璃量器和移液器、天平,并适当增加标准工作液的测定次数和平行样品的测定,以降低整个检测过程中可能产生的不确定度,提高检测结果的准确性。
[1] 祝伟霞,刘亚风,梁炜. 动物性食品中硝基呋喃类药物残留检测研究进展[J]. 动物医学进展, 2010, 31(2): 99-102.
[2] 唐红梅,曾芳,李成洪. 食品中硝基呋喃类药物及其代谢物残留检测的研究进展[J].食品安全质量检测学报, 2016, 7(10):3952-3959.
[3] 王群,马兵,吕海燕,等.食品中硝基呋喃类及其代谢物对人体健康的安全性评价[J].中国渔业质量与标准, 2013, 3(2):4-10.
[4] 蒋原,丁涛,徐锦忠. 硝基呋喃类药物在克氏鳌虾组织中消除规律的研究[J]. 畜牧与兽医, 2008, 40(2):34-37.
[5] Hoogenboom L A,van Bruchem G D, et al. Absorption of amutagenic metabolite released from protein-bound residues of furazolidone[J]. Environ Toxicol Pharmacol, 2002,11(3): 273-287.
[6] European Food Safety Authority.Opinion of the scientific panel on food additives, flavourings, processing aids and materials in contact with food on a request from the Commission related to Semicarbazide in food[J].EFSA J, 2005, 219:1-36.
[7] Mottier P, Khong S P, Gremaud E, et al.Quantitative determination of four nitrofuran metabolites in meat by isotope dilution liquid chromatography-electrospray ionization-tandem mass spectrometry[J]. J Chromatogr A, 2005, 1069(1/2):85-91.
[8] 中华人民共和国农业部.中华人民共和国农业部公告第235号[EB/OL]. (2002-12-24) [2017-03-05].http://www.moa.gov.cn/zwllm/nybz/200803/t20080304_1028649.htm.
[9] 中华人民共和国农业部.中华人民共和国农业部公告第560号[EB/OL].(2005-10-28) [2017-03-05]. http://www.moa.gov.cn/zwllm/tzgg/gg/200511/t20051117_496523.htm.
[10] 中华人民共和国卫生部.关于印发《食品中可能违法添加的非食用物质和易滥用的食品添加剂名单(第四批) 》的通知[EB/OL]. (2010-03-22) [2017-03-05].http://www.scfda.gov.cn/CL2488/77930.html.
[11] 柳爱春,刘超,赵芸,等.免疫胶体金法快速检测水产品中硝基呋喃类代谢物的研究[J].浙江农业学报, 2013, 25(1):95-102.
[12] 蒋宏伟. 酶联免疫技术在动物产品中硝基呋喃类药物残留检测的应用[J].陕西农业科学, 2006(5):53-55.
[13] 王媛,蔡友琼,贾东芬,等.高效液相色谱法检测水产品中硝基呋喃类代谢物残留量[J].分析试验室, 2009, 28(12):86-90.
[14] 李绪鹏,彭家杰,胡浩光,等.超高效液相色谱串联质谱法测定水产品中硝基呋喃类代谢物的不确定度分析[J].农业与技术, 2016, 36(23):1-4.
[15] 祝子铜,雷美康,彭芳,等.液质联用测定水产品中硝基呋喃类代谢物残留量的不确定度评定[J].食品安全质量检测学报, 2015, 6(7):2857-2862.
[16] Cordero R, Seckmeyer G, Pissulla D,et al. Uncertainty evaluation of the spectral UV irradiance evaluated by using the UVSPEC radiative transfer model[J]. Opt Commun, 2007, 276(12): 44-53.
[17] 全国认证认可标准化技术委员会.GB/T 27025—2008:检测和校准实验室能力的通用要求[S].北京:中国标准出版社, 2008.
[18] 中国合格评定国家认可委员会. 化学分析中的不确定度评估指南[M]. 北京:中国计量出版社, 2002.
[19] 国家质量监督检验检疫总局.JJF 1059.1—2012测量不确定度的评定与表示[S]. 北京: 中国计量出版社,2012.
[20] 常晨阳,王碧生,方成俊,等. LC-MS/MS法测定动物源食品中呋喃西林代谢物不确定度的评定[J].现代农业科技, 2014(12):274-276.
[21] 陈国花. LC-MS/MS法测定动物源性食品中呋喃它酮代谢物试验[J].中国畜牧兽医文摘,2014,30(8):46.
[22] 何太喜,段兵,张文. 液相色谱-串联质谱法测定鳗鱼肉中硝基呋喃类药物代谢物残留量不确定度分析[J]. 分析试验室, 2009, 28:281-284.
[23] 国家质量监督检验检疫总局.GB/T 21311—2007 动物源性食品中硝基呋喃类药物代谢物残留量检测方法高效液相色谱/串联质谱法[S]. 北京: 中国计量出版社, 2007.
[24] 邢丽红,付树林,孙晓杰,等.液相色谱-串联质谱法测定大菱鲆中硝基呋喃类代谢物的不确定度评定[J].现代农业科技, 2015(7):293-296.
[25] 郭丽娜,刘成文,符元春.液相色谱串联质谱法测定水产品中硝基呋喃类代谢物的不确定度评定[J]. 现代食品, 2017(2):101-105.
[26] 中华人民共和国农业部.中华人民共和国农业部公告第783号[EB/OL]. (2008-03-21) [2017-03-05]. http://www.moa.gov.cn/zwllm/nybz/200803/t20080321_1028664.htm.
[27] 国家质量监督检验检疫总局.JJG196—2006 常用玻璃量器检定规程 [S]. 北京: 中国计量出版社, 2006.
[28] 张向宇. 实用化学手册[M]. 第2版.北京:国防工业出版社,2011.
[29] 国家质量监督检验检疫总局.JJG646—2006 移液器检定规程 [S]. 北京: 中国计量出版社, 2006.
Uncertaintyevaluationindeterminationofnitrofuranmetabolites residuesintilapiabyUHPLC-MS/MS
LINGongshi
(XiamenMarine&FisheriesResearchInstitute,Xiamen361008,China)
In order to determine the main source of uncertainty of nitrofuran metabolites residues in tilapia, according to “Evaluation and expression of uncertainty in measurement (JJF1059.1-2012)” and other related theories, the study constructed a corresponding mathematical model for nitrofuran metabolites residues in tilapia based on the method of sample pretreatment and calculation formula from the announcement of Ministry of Agriculture (No.783-1-2006). When the spiked level was1.00μg/kg, the results of4nitrofuran metabolites were showed thatCSEMwas (0.989±0.168) μg/kg,k=2;CAHDwas (1.009±0.165) μg/kg,k=2;CAMOZwas (0.991±0.169) μg/kg,k=2; andCAOZwas (0.986±0.180) μg/kg,k=2. It suggested that the uncertainty were mainly from the preparation of calibration solution and internal standard solution, linear curve fitting, and the spiked recovery under determined experimental conditions. The study was of great significance on reducing uncertainty for detection of nitrofuran metabolites residues and ensuring accuracy and reliability of test data. [Chinese Fishery Quality and Standards,2017,7(6):48-57]
ultra high performance liquid chromatography-tandem mass spectrometry(UHPLC-MS/MS); tilapia; nitrofuran metabolites residues; uncertainty
LIN Gongshi, lgsqz@163.com
《中国食物与营养》2018年征稿征订启事
中国科技核心期刊 中国农业核心期刊
《中国食物与营养》创办于1995年,由农业部主管,中国农业科学院、国家食物与营养咨询委员会主办的食物与营养领域相结合的综合性月刊,国内外公开发行。2014年影响因子为0.937。
办刊宗旨:立足于农业、食物、营养领域的结合,报道国家在食物与营养相关领域的方针、政策、法规、标准等;刊登食物生产、食物消费、食品工业、食物营养、公共营养、临床营养等方面的发展动态和科技成果等。
本刊主要栏目有:专题论坛、食物安全、资源与生产、食品工业、消费与流通、新技术新产品、营养与保健、营养与疾病、膳食营养调查等。
欢迎大家踊跃投稿和订阅《中国食物与营养》杂志。
《中国食物与营养》杂志由北京报刊发行局发行,邮发代号为82-597。本刊为月刊,每期定价15元,全年180元。也可直接汇款到编辑部订阅(免费邮寄)。
地址:北京市海淀区中关村南大街12号《中国食物与营养》编辑部
电话:(010)82109761传真:(010)82106285邮编:100081
在线投稿系统:http://foodandn.caas.cn
E-mail:foodandn@263.net;sfnccli@163.com
10.3969/j.issn.2095-1833.2017.06.008
S98
A
2095-1833(2017)06-0048-10
2017-03-28;接收日期2017-10-19
厦门市水产品质量安全监测专项(506007)
林功师(1985-),男,硕士,工程师,研究方向为水产品质量安全检测与研究,lgsqz@163.com
猜你喜欢
化工管理(2022年13期)2022-12-02
世界农药(2021年3期)2021-12-09
化工管理(2021年7期)2021-05-13
中国食品(2020年9期)2020-05-26
农药科学与管理(2019年12期)2019-05-20
中成药(2018年1期)2018-02-02
浙江化工(2015年1期)2015-11-24
中国药业(2014年24期)2014-05-26
火炸药学报(2014年5期)2014-03-20
火炸药学报(2014年5期)2014-03-20
