
基于RNA-seq技术的宫颈癌与癌旁组织差异表达基因分析
2017-12-27 08:09张志珊蒋燕成陈紫萱陈婉花李爱禄李纯孝
实用癌症杂志 2017年12期
张志珊 蒋燕成 陈紫萱 陈婉花 李爱禄 李纯孝
·基础研究·
基于RNA-seq技术的宫颈癌与癌旁组织差异表达基因分析
张志珊 蒋燕成 陈紫萱 陈婉花 李爱禄 李纯孝
目的探讨宫颈癌的发病机制。方法应用RNA-seq技术分析3对宫颈癌及癌旁组织转录组,利用生物信息方法对差异基因进行基因本体(GO)分析和通路富集性分析。结果在宫颈癌组织中发现1755个差异表达基因,其中758个基因表达上调,997个基因表达下调。功能富集分析表明,差异基因富集于细胞粘附、DNA损伤有关的信号通路上。且宫颈癌组织中发现RAB22A-BCAS1等融合基因。结论细胞粘附信号通路、RAB22A-BCAS1融合基因可能与宫颈癌的发生机制有关。
宫颈癌;RNA-seq;转录组;差异表达基因;融合基因
宫颈癌是女性最常见的恶性肿瘤之一,人乳头状瘤病毒(HPV)的感染是引起宫颈癌的重要因素,但并非所有感染HPV的患者都会发展为宫颈癌,只有HPV持续感染导致细胞的基因组异常才最终导致宫颈癌的发生。本研究拟收集福建医科大学附属泉州第一医院妇科就诊的宫颈癌患者3例,采用RNA-Seq技术分析宫颈癌组织与癌旁组织的转录组,筛选出差异表达基因,进而分析差异表达基因所涉及的信号通路以及融合基因表达,有利于阐明宫颈癌的发病机制,寻找宫颈癌特异的分子标志物,同时也为基因药物的开发提供候选的药物作用靶点。
1 材料与方法
1.1 研究对象
收集福建医科大学附属泉州第一医院妇科就诊的宫颈癌患者3例,术前经未经放化疗及药物治疗,经病理组织学确诊为宫颈癌ⅡB期,同时取癌组织和癌旁组织(距离宫颈癌癌灶边缘>3 cm),置于RNA保存液中-80 ℃保存。
1.2 RNA提取及质检
3对癌组织及癌旁组织分别用QIAGEN微量RNA提取试剂盒按操作说明书提取RNA。琼脂糖凝胶电泳分析 RNA 的纯度和完整性,Nanodrop 检测 RNA 的纯度(OD 260/280 比值),Qubit 2.0对RNA浓度进行精确定量,Agilent 2100精确检测RNA的完整性。
1.3 文库构建及测序
用带有Oligo(dT)的磁珠富集真核生物 mRNA,加入 fragmentationbuffer 将 mRNA 打断成短片段,以 mRNA 为模板,用六碱基随机引物(random hexamers)合成一链 cDNA,加入缓冲液、dNTPs 和 DNA polymerase I 和RNase H 合成二链 cDNA,利用 AMPure XP beads 纯化双链 cDNA。纯化的双链 cDNA 先进行末端修复、加 A 尾并连接测序接头,再用 AMPure XPbeads 进行片段大小选择。采取PCR 扩增,用 AMPure XP beads 纯化 PCR 产物,得到文库。文库构建完成后,先使用 Qubit 2.0 进行初步定量,稀释文库,使用 Agilent 2100 对文库的插入片段进行检测,使用 Q-PCR 方法对文库的有效浓度进行准确定量,保证文库质量。文库检测合格后,使用 Illumina 高通量测序平台(HiSeq/MiSeq)进行测序。
1.4 测序数据分析
测序数据的质量主要分布在Q30以上,才能保证后续高级分析的正常进行。将原始数据中包含的接头(adapter)信息、低质量碱基、未测出的碱基去除,得到最终的有效数据(clean data)。利用TopHat2软件将获得的数据与参考基因组进行序列比对,比对到指定的参考基因组上的Reads称为Mapped Reads,利用Cufflinks软件将Mapped Reads 进行转录本拼接,利用Cufflinks软件计算癌组织和癌旁组织基因表达量。
1.5 差异表达基因筛选
应用CuffDiff软件进行分析,在不同样本组差异表达RNA检测过程中,将Fold Change≥2且FDR<0.05作为筛选标准。差异倍数(Fold Change)表示两样品间表达量的比值。
1.6 差异表达基因通路富集及基因本体(GO)分析
应用信号通路分析软件(IPA)对2组样本差异表达基因进行信号通路富集、疾病功能富集分析,利用软DAVID工具进行GO分析。
1.7 融合基因分析
应用TopHat fusion分析融合基因,tophat fusion通过将未能比对到参考基因组的reads打碎成25 bp的片段,然后采用Bowtie将25 bp的片段比对到基因组,从而找出比对到两个染色体的片段或者比对到同一染色体不同位置的reads。
2 结果
2.1 差异表达基因分析
将癌组织和癌旁组织相比,共发现1755个差异表达基因,癌组织相对于癌旁组织样本中有758个基因表达量上调,997个基因表达量下调。
2.2 GO及通路分析
1755个差异基因共富集到582条GO_BP,1755个差异表达基因共富集到378条通路。这些差异表达的基因主要涉及粘附、DNA损伤等通路。最有意义的前5条信号通路富集结果如表1所示。

表1 癌组织和癌旁组织差异表达基因前5条信号通路富集结果
2.3 融合基因
分析癌组织中存在的融合基因,分析结果及说明见表2。
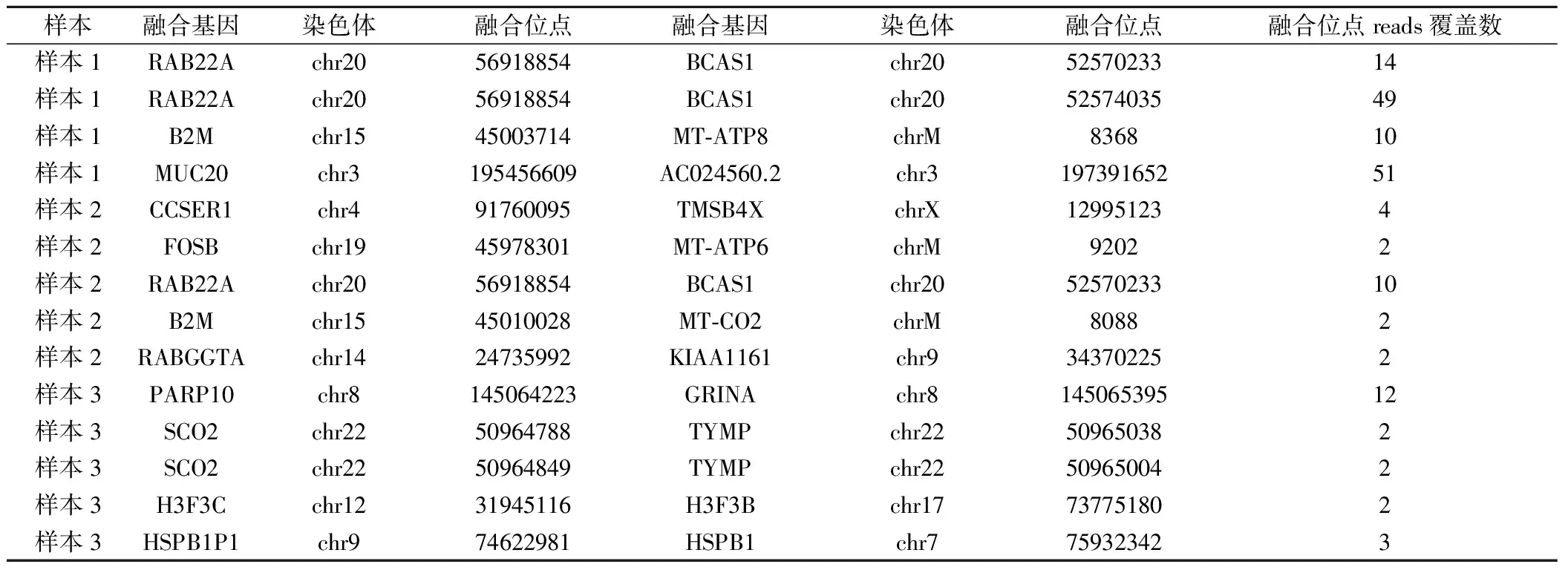
表2 融合基因分析结果及说明
3 讨论
虽然高危型HPV的持续感染是引起宫颈癌的根本原因,但仅有一小部分感染者发展为宫颈癌[1]。目前众多研究证实宫颈组织基因水平上表达异常会诱发宫颈癌的发生[2]。RNA-Seq技术不仅能检测组织已知基因的转录活动,还能检测未知基因[3]。
本研究中收集福建医科大学附属泉州第一医院妇科就诊的宫颈癌患者3例,采取RNA-Seq技术,共发现1755个差异表达基因,共富集到582条GO种类和378条通路,这些差异表达的基因主要涉及粘附、DNA损伤等通路。排在前2位的通路均与细胞粘附和渗出有关,而细胞粘附分子在细胞的粘附与渗出中起重要的作用。研究显示细胞粘附分子与宫颈癌的侵袭转移有密切关系。在宫颈癌的形成、发展和转移过程中,细胞粘附分子起着至关重要的作用[4]。活化白细胞粘附分子在不同恶变程度宫颈癌中的表达存在差异,并在宫颈癌的发生发展过程中表达量呈上升趋势[5]。因此我们推测与细胞粘附相关的信号通路可能在宫颈癌的发生中起一定的作用。
融合基因是指两个基因的编码区首尾相连构成的嵌合基因。融合基因编码的蛋白通常具有致癌性,会影响细胞的正常生理功能,是导致癌症的主要原因之一。目前,在肺癌、甲状腺、乳腺癌等疾病中,都发现了融合基因的存在[6-7],但是与宫颈癌的相关性尚未见报道。本研究通过RNA-Seq技术检测宫颈癌组织和癌旁组织的转录组发现,与癌旁组织相比,3个宫颈癌组织中都存在不同程度的融合基因。其中RAB22A-BCAS1融合基因有2个宫颈癌样本中同时存在。RAB22A是ras相关的小G蛋白家族成员,是囊泡运输重要的调节因子。RAB22A在包括肝癌、黑色素瘤、卵巢癌、骨肉瘤在内的多种肿瘤中过表达[8-9]。乳腺癌扩增序列-1 (BCAS1) 位于20q13.2区域,BCAS1高表达在乳腺癌浸润和转移中发挥重要的作用[10],BCAS1还参与三阴乳腺癌的发生[11]。此外,有研究表明,BCAS1还与前列腺癌、胃食管连接处腺癌、胰腺癌相关[12]。RAB22A、BCAS1与宫颈癌的相关性尚未见报道,而RAB22A-BCAS1融合基因以及本研究中发现的其他融合基因是否为导致宫颈癌的潜在致癌基因,在后续的研究中,我们将扩大样本量进行进一步的验证。
综上所述,我们通过高通量RNA-Seq技术分析宫颈癌及癌旁转录组,发现1755个差异表达基因,这些差异表达基因富集到与细胞粘附、DNA损伤等信号通路上,且发现了RAB22A-BCAS1等融合基因,推测可能与宫颈癌的发生、发展相关,为后续进一步研究宫颈癌的发生机制提供了新的方向和思路。
[1] Moody CA,Laimins LA.Human papillomavirus oncoproteins:pathways totransformation〔J〕.Nat Rev Cancer,2010,10(8):550-560.
[2] Ojesina AI,Lichtenstein L,Freeman SS,et al.Landscape of genomic alterations in cervical carcinomas〔J〕.Nature,2014,506 (7488):371-375.
[3] Maher CA,Kumar-Sinha C,Cao X,et al.Transcriptome sequencing to detect gene fusions in cancer〔J〕.Nature,2009,458(7234):97-101.
[4] 刘成红,毛熙光.细胞粘附分子与宫颈癌的侵袭转移〔J〕.西南军医,2008,10(5):95-97.
[5] 杨 潇,王艳清,鲜舒等.活化白细胞黏附分子在宫颈癌中的表达及功能研究〔J〕.医学分子生物学杂志,2016,13(5):267-271.
[6] Wu YM,Su F,Kalyana-Sundaram S,et al.Identification of targetable FGFR gene fusions in diverse cancers〔J〕.Cancer Discov,2013,3(6):636-647.
[7] Panagopoulos I,Gorunova L,Bjerkehagen B,et al.Chromosome aberrations and HEY1-NCOA2 fusion gene in a mesenchymal chondrosarcoma〔J〕.Oncol Rep,2014,32(1):40-44.
[8] Zhou Y,Wu B,Li JH,et al.Rab22a enhances CD147 recycling and is required for lung cancer cell migration and invasion〔J〕.Exp Cell Res,2017,357(1):9-16.
[9] Su F,Chen Y,Zhu S,et al.RAB22A overexpression promotes the tumor growth of melanoma〔J〕.Oncotarget,2016,7(44):71744-71753.
[10] Tanner MM,Tirkkonen M,Kallioniemi A,et al.Amplification of chromosomal region 20q13 in invasive breast cancer:prognostic implications〔J〕.Clin Cancer Res,1995,1(12):1455-1461.
[11] Zeitz MJ,Ay F,Heidmann JD,et al.Genomic interaction profiles in breast cancer reveal altered chromatin architecture〔J〕.PLoS One,2013,8(9):e73974.
[12] Loukopoulos P,Shibata T,Katoh H,et al.Genome-wide array-based comparative genomic hybridization analysis of pancreatic adenocarcinoma:identification of genetic indicators that predict patient outcome〔J〕.Cancer Sci,2007,98(3):392-400.
AnalysisofDifferentlyExpressedGenesofTumorandAdjacentNon-tumorTissuesfromPatientswithCervicalCancerbyRNASequencing
ZHANGZhishan,JIANGYancheng,CHENZixuan,etal.
QuanzhouFirstHospital,FujianMedicalUniversity,Quanzhou,362000
ObjectiveTo explore the possible pathogenesy of cervical cancer.MethodsRNA sequencing was performed to screen the differently expressed genes of 3 pairs of cervical carcinoma and matched adjacent non-tumor tissues.The differently expressed genes were identified with gene ontology (GO) analysis and pathway enrichment analysis.ResultsThere were a total of 1755 differently expressed genes in the samples,including 758 up-regulated genes and 997 down-regulated genes.These differently expressed genes were enriched in pathway related to cell adhesion and DNA damage.Moreover,RAB22A-BCAS1 fusion gene was found in cervical cancer tissues.ConclusionThe pathway of cell adhesion and RAB22A-BCAS1 fusion gene may be related to the tumorigenesis and development of cervical cancer.
Cervical carcinoma;RNA-seq;Transcriptome;Differently expressed gene;Fusion gene
(ThePracticalJournalofCancer,2017,32:1915~1917)
福建省自然科学基金(编号:2014J01436)
362000 福建医科大学附属泉州第一医院
10.3969/j.issn.1001-5930.2017.12.001
R737.33
A
1001-5930(2017)12-1915-03
2017-06-22
2017-07-10)
(编辑:吴小红)
猜你喜欢
中老年保健(2021年12期)2021-08-24
猪业科学(2021年3期)2021-05-21
传染病信息(2021年6期)2021-02-12
中国生殖健康(2020年7期)2021-01-18
幽默大师(2020年10期)2020-11-10
中华诗词(2019年1期)2019-11-14
科海故事博览·下旬刊(2019年6期)2019-04-16
中国生殖健康(2018年4期)2018-11-06
猪业科学(2018年4期)2018-05-19
河北医学(2016年5期)2016-12-01
