
碱改性HZSM-5催化热解木质素催化剂失活分析
2017-12-22 05:37唐松山泮泽优张长森王登台薛翔飞曹云峰刘永刚张瑞芹
化工学报 2017年12期
唐松山,泮泽优,张长森,王登台,薛翔飞,曹云峰,刘永刚,张瑞芹
(1郑州大学化学与分子工程学院,河南 郑州 450001;2 河南省环境化学与低碳技术重点实验室,河南 郑州 450001;3天方药业有限公司,河南 驻马店 463000)
碱改性HZSM-5催化热解木质素催化剂失活分析
唐松山1,3,泮泽优1,2,张长森1,2,王登台1,2,薛翔飞1,2,曹云峰3,刘永刚1,2,张瑞芹1,2
(1郑州大学化学与分子工程学院,河南 郑州 450001;2河南省环境化学与低碳技术重点实验室,河南 郑州 450001;3天方药业有限公司,河南 驻马店 463000)
对0.3 mol·L-1NaOH改性后的HZSM-5以及未改性HZSM-5催化剂进行循环和再生评价实验以考察催化剂的寿命。对反应后和再生的催化剂进行N2吸脱附以及NH3-TPD表征,并通过对反应后的催化剂进行SEM、TGA、FTIR、UV-Vis等表征分析催化剂积炭。两种催化剂的活性均随着循环实验次数的增加而逐渐降低,经4次循环实验的改性HZSM-5催化剂的催化活性远高于4次循环实验的未改性HZSM-5催化剂。反应过的催化剂经高温煅烧再生后其活性都有所恢复,再生后的改性HZSM-5的催化活性仍高于未改性催化剂。对两种不同催化剂积炭分析,改性后催化剂的积炭量少于未改性的催化剂,其积炭组分中高聚芳烃的含量相对较多。
HZSM-5分子筛;催化热解;木质素;失活;再生;积炭
引 言
快速催化热解(catalytic fast pyrolysis,CFP)是一种将木质素转化为芳烃类化合物等具有高附加值化学品的资源化利用方式[1-7]。对于木质素的快速催化热解,目前比较有效的催化剂是具有强酸性和良好择形催化功能的HZSM-5系列的催化剂[1-4,8-10]。由于孔道口直径的限制,木质素热解生成的愈创木酚(三维尺寸 0.9476 nm × 0.8101 nm × 0.4197 nm)和紫丁香醇(三维尺寸1.0738 nm × 0.7856 nm ×0.4218 nm)很难进入到HZSM-5催化剂的孔道(0.55 nm × 0.51 nm & 0.56 nm × 0.53 nm)内参与催化反应[9],只能停留在催化剂的外表面,这些物质在催化剂的外表面很难得到有效转化,容易聚集结焦生成焦炭,堵塞催化剂孔道,导致催化剂失活。为了提高木质素热解生成的大分子含氧物质的转化率,具有微孔和介孔结构的多级孔HZSM-5被认为是一种非常有前景的催化剂[9,11],木质素热解生成的大分子含氧物质能进入到介孔内参与催化转化,同时该类催化剂又具有微孔结构的芳构化能力[9,11-13]。
Du等[14]研究HZSM-5分子筛在催化转化生物质热解油模型化合物过程中催化剂积炭的问题,指出产物中高价值化学品选择性下降并且催化剂活性降低主要归因于催化剂孔道堵塞以及催化剂活性位点中毒。尹海云等[15]分析了HZSM-5 催化剂在线提质生物油的失活机理,在催化反应过程中,催化剂孔道内生成的石墨状积炭及在表面形成的纤维状积炭覆盖了催化剂的活性位点,使得催化剂活性降低。在HZSM-5 催化提质生物质热解油过程中,催化剂的弱酸性位点上主要发生裂解和异构化反应,强酸性位点上主要发生芳构化反应[16],随着催化剂使用时间的延长,强酸位点逐渐被积炭覆盖致使强酸酸性减弱,使得催化剂活性下降[15]。对于HZSM-5分子筛催化剂催化热解木质素,木质素热解生成的酚类化合物会吸附在催化剂的活性位点上导致催化剂活性降低,并且会有积炭产生[2,17]。
然而针对于实际工业化生产,催化剂价格高昂,并且催化剂需求量大,因此在连续不断的工业生产中需对催化剂进行多次循环使用。基于此,研究催化剂的失活、再生以及再利用等对于催化剂的工业化应用具有十分重要的意义。
从之前的研究工作[13,18-19]可以看出,碱改性能制备出具有微孔和介孔的多级孔HZSM-5分子筛催化剂,并且0.3 mol·L-1NaOH的改性效果最好。很少有人研究HZSM-5以及脱硅改性后的HZSM-5催化热解木质素催化剂的失活与再生,因此在本研究中主要考察了 HZSM-5分子筛以及 0.3 mol·L-1NaOH改性后的HZSM-5分子筛催化热解木质素的失活与再生问题,并对反应后催化剂进行了催化积炭分析。
1 实验材料和方法
1.1 实验药品及试剂
HZSM-5分子筛购买于南开大学催化剂厂,NaOH(AR)由天津市科密欧试剂厂提供,木质素(CAS# 8068-05-1,BR)购买于上海源叶生物科技有限公司。
1.2 实验方法
改性催化剂制备:将HZSM-5置于马弗炉中550℃焙烧2 h,备用。取50 ml的0.3 mol·L-1NaOH溶液置于圆底烧瓶内,水浴加热到70℃,称取5 g焙烧后的分子筛加入到烧瓶内,磁力搅拌、回流2 h,取出烧瓶在冷水浴中迅速冷却,洗涤、抽滤3次,将滤饼 110℃干燥过夜。将干燥后的样品在 1 mol·L-1NH4NO3溶液中 80℃水浴磁力搅拌回流 2 h,重复3次,抽滤,滤饼110℃干燥过夜。550℃焙烧干燥后样品3 h,即制得改性催化剂。
催化热解实验在 BTF-1200C-RTP型(Anhui BEQ Equipment Technology Co.,Ltd)快速升温管式炉上进行,将1 g木质素和1 g催化剂装入样品管(长500 mm,内径16 mm),木质素和催化剂由石英棉隔开,确保木质素热解蒸汽全部穿过催化剂床层。通入100 ml·min-1的高纯氮气10 min置换装置内的空气,以100℃·s-1的升温速率升至600℃热解1min,液体产物采用冰水浴冷凝收集,气体产物用集气袋收集。取出反应后的催化剂,在 CDS Pyroprobe 5200HP-R型裂解仪(CDS Analytical Inc.)上以木质素为原料进行评价实验,裂解产物由 GC/MS(Shimadzu QP2010 Ultra)检测分析。
1.3 分析与测试
木质素在 CDS仪器内的裂解产物在氦气的带动下经由传输线注入到GC/MS系统进行在线分析。GC/MS进样口温度250℃,1 ml·min-1高纯氦气为载气,色谱柱型号 RXI-5SiL-MS(30.0 m×0.25 mm×0.25 μm),分流比 100:1,升温程序:50℃保持3 min,以4℃·min-1升至200℃后保持10 min,离子源温度200℃,70 eV的EI离子化方式,质谱扫描范围m/z:33~550 amu,2000 amu·s-1。采用NIST11质谱库匹配质谱检出物,采用峰面积归一法计算各类化合物的相对含量,以烃类产物的选择性作为评价指标。
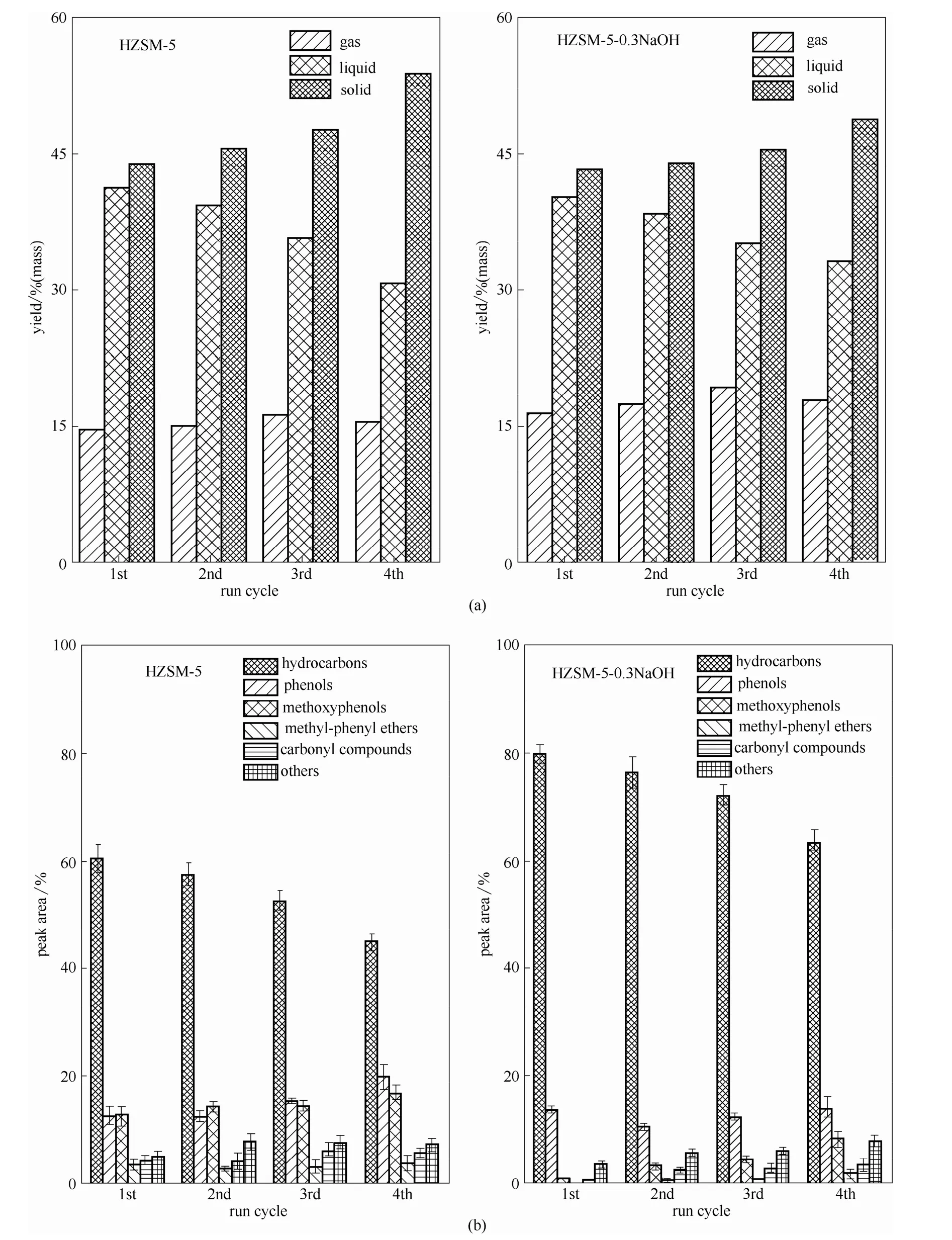
图1 两种催化剂循环实验产率及各产物选择性Fig.1 Yields(a) and selectivity(b) of HZSM-5 and modified HZSM-5 cyclic tests
催化剂的 N2等温吸脱附曲线以及孔道结构参数在ASAP2420-4MP(Micromeritics,USA)型全自动比表面仪上获得,样品测试之前300℃脱气2 h。催化剂的 NH3-TPD曲线在 Autosorb-iQ(Quantachrome,USA)型全自动气体吸附仪上测得,对NH3脱附曲线进行峰面积积分,换算出催化剂的酸性量。催化剂的SEM图片在Hitachi S-4800(HITACHI,Japan)型扫描电镜上拍得。采用STA 449 F3(Netzsch,Germany)同步热分析仪对样品进行TGA分析。采用NEXUS 470(Niclet,UAS)型红外光谱仪对样品进行红外光谱分析,溴化钾压片法。采用Cary 5000(Agilent,UAS)型分光光度计对样品进行UV-Vis分析,波长范围200~800 nm。
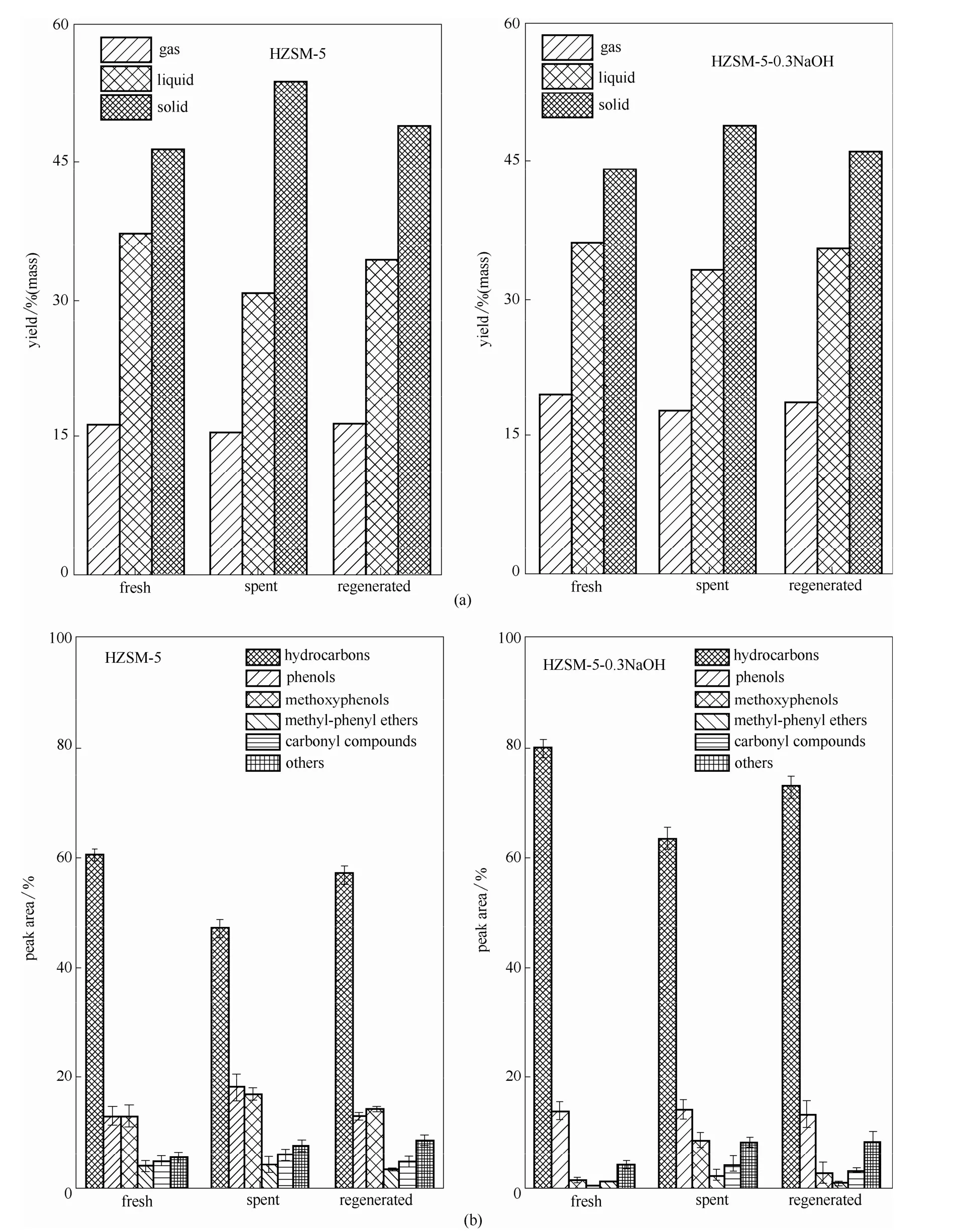
图2 两种催化剂再生实验产率及各产物选择性Fig.2 Yields(a) and selectivity(b) of HZSM-5 and modified HZSM-5 regeneration test
2 结果与讨论
2.1 催化剂寿命考察
2.1.1 循环实验 从图1可以看出对于两种不同的催化剂,随着循环实验次数的增加,液体产率逐渐减少,固体产率逐渐增加,气体产率的变化不是很明显。对于两种催化剂,其热解生成烃类产物的选择性都是随着催化循环实验次数的增加而逐渐降低,两种不同催化剂在经4次循环实验后其活性都有所降低。不同之处在于进行4次循环实验后,产物中烃类产物选择性降低程度不同,未改性HZSM-5经循环实验后其GC/MC检测产物中烃类产物的选择性降至 57%;0.3 mol·L-1NaOH 改性HZSM-5经4次循环实验后其产物中烃类产物的选择性降至72%。
从中可以看出,经过循环实验后,0.3 mol·L-1NaOH改性 HZSM-5的催化活性经循环实验后降低,但其催化热解木质素生成烃类产物的效果仍比未改性HZSM-5的催化效果好。
2.1.2 再生实验 催化剂的再生处理为取出循环实验后的催化剂置于马弗炉中550℃煅烧1 h,以确保催化剂积炭被燃烧完全。从图2中可以看出,对于两种催化剂,再生催化剂催化液体产率均低于新鲜催化剂催化液体产率,高于反应后催化剂催化热解木质素的液体产率;热解液体产物中烃类产物的选择性水平都有所恢复,但均不能恢复到新鲜催化剂的水平。这表明反应后催化剂进行催化热解实验时其活性降低,通过燃烧再生的方法能使得催化剂的活性恢复,但是不能恢复到新鲜催化剂的水平。
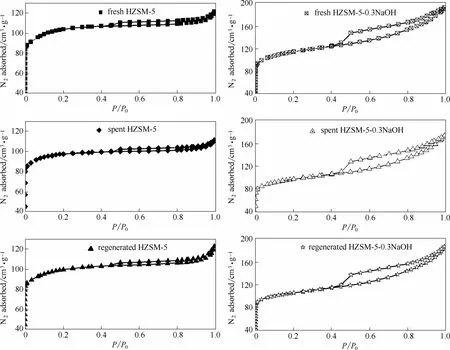
图3 反应前、后及再生催化剂的N2吸脱附曲线Fig.3 N2 adsorption/desorption isotherms of fresh,spent and regenerated catalyst samples
不同之处在于二者催化剂活性水平恢复能力不同,未改性的催化剂反应后经燃烧再生后,其热解产物中烃类选择性水平恢复至新鲜催化剂的95%;再生的0.3 mol·L-1NaOH改性HZSM-5催化剂热解产物中烃类选择性水平恢复至新鲜催化剂的90%。可以看出改性催化剂的再生活性恢复能力不及未改性催化剂活性恢复能力强,这主要是由于碱改性对催化剂的晶体结构有一定损害,在催化反应以及催化剂再生过程中晶体所受到的损害会进一步加大,因此再生后的催化活性水平恢复能力要稍弱于未改性的催化剂。
2.1.3 N2吸脱附表征 从图3中可以看出反应后以及再生的HZSM-5和改性HZSM-5吸附等温线与新鲜催化剂一样仍分别呈现为Ⅰ型和Ⅳ型等温线,并且反应后以及再生后的改性HZSM-5催化剂的等温线与新鲜改性HZSM-5催化剂一样呈现H4型滞后环,结合表1的数据可以看出反应后催化剂的比表面积、孔容等减少,经再生处理后其比表面积、孔容等能基本恢复至新鲜水平,这表明催化剂经再生处理后能基本恢复原有的孔道水平。在木质素热解过程中会产生两种不同的催化剂积炭:一种是木质素热解生成的中间体发生二次聚合反应而生成的存在于催化剂晶粒的表面以及不同晶粒之间比较大的空隙内的积炭[20],存在于催化剂晶体表面的积炭很容易堵塞催化剂的孔道口;另外一种积炭是木质素热解生成的中间体进入到催化剂的孔道内在催化剂的酸性位点上发生反应而生成的积炭[4,21-22]。因此反应后催化剂的比表面积、孔容等会有损失,再生处理能将积炭燃烧掉,这样会在一定程度上恢复催化剂的比表面积、孔容等。
对于未改性催化剂催化热解木质素,由于催化剂孔道口径的限制,木质素热解生成的大分子反应中间体很难进入到催化剂的孔道内,会在催化剂的孔道口聚集生成积炭进而堵塞催化剂孔道口,那些生成的小分子的中间产物很难再进入到催化剂的孔道内参与催化反应,因此其有效孔容就会相应地降低;而对于改性后的催化剂催化热解木质素,生成的大分子中间体不再受催化剂孔道口径的限制,很容易地就能够进入到其孔内参与催化反应,不会再堵塞催化剂的孔道口,因此其孔容损失量相对要少。由于改性催化剂是以牺牲催化剂结构的完整性为代价的,因此改性后催化剂在经历反应以及再生等过程后,其结构的完整性会进一步遭到破坏,因此再生后其孔容的恢复能力不及未改性催化剂的恢复能力强。
2.1.4 NH3-TPD表征 从图4中可以看出反应后以及再生后的催化剂与未参与反应的催化剂同样具有弱酸峰和强酸峰。
从图5中可以看出,反应后两种催化剂无论是弱酸量还是强酸量都有所下降。在木质素催化热解反应过程中,木质素热解中间体在催化剂酸性位点上发生催化反应,会对催化剂的酸性结构造成一定的不可逆性改变,因此反应后催化剂的酸性量会有所降低。碱改性是以脱除分子筛骨架原子为代价增加催化剂孔径水平的,改性后的催化剂酸性位点的结构遭到损坏,因此在参与催化反应后其酸性位点保持原有结构的能力不如未改性催化剂的能力强,所以反应后未改性HZSM-5的酸量降低的程度要小于反应后的改性HZSM-5的酸量降低程度。反应后的催化剂再生后其酸性水平会有所恢复,但是不能恢复到原有酸性水平。催化剂的再生主要是通过高温煅烧的方式烧掉催化剂积炭,那些与催化剂酸性位点进行可逆性结合的积炭会被烧掉,因此催化剂的酸性水平会有一定程度的恢复;但高温煅烧无法恢复失活酸性位点的原有结构,因此经过再生后催化剂酸性不能恢复到原有的水平。
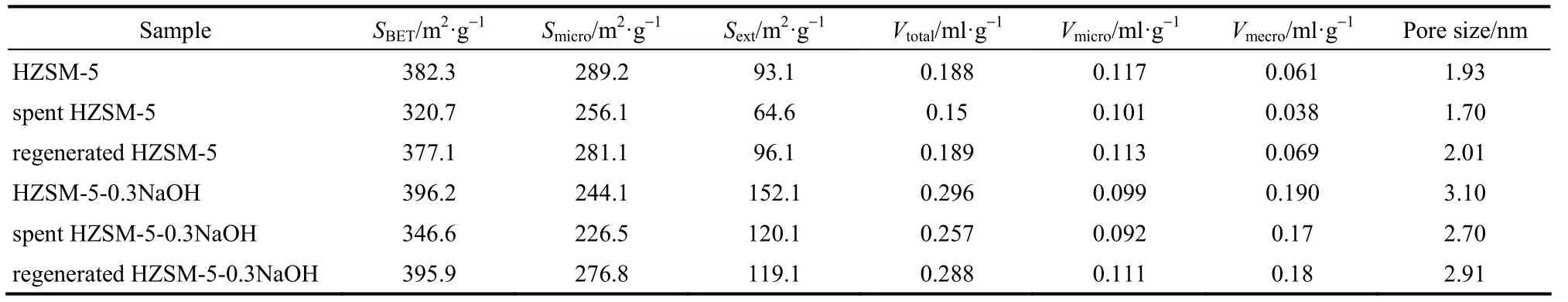
表1 反应前、后及再生催化剂的孔结构参数Table 1 Porous properties of catalyst samples
从 N2吸脱附和 NH3-TPD的分析结果可以看出,反应后的催化剂因积炭堵塞孔道和催化剂活性位点中毒(酸性结构发生不可逆性改变)而失活。催化剂积炭可以通过高温煅烧的方式除去,而催化剂酸性结构的不可逆性改变很难再恢复,因此在后面的分析中不再考虑催化剂的活性位点中毒,只对催化剂积炭进行分析。
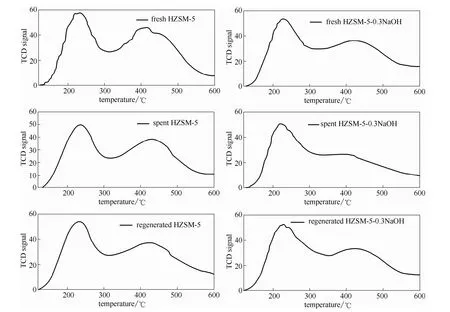
图4 反应前、后及再生催化剂的NH3-TPD曲线Fig.4 NH3-TPD curves of catalyst samples

图5 反应前、后及再生催化剂的酸量Fig.5 Concentration of acid sites of fresh,spent and regenerated catalyst samples
2.2 反应后催化剂积炭表征分析
2.2.1 SEM表征 从图6中可以看出,新鲜催化剂的表面比较干净,没有多余的杂质,新鲜HZSM-5催化剂[图 6(a)]的晶体结构相对比较完整,改性后的催化剂[图 6(b)]由于碱溶液脱除了一部分骨架原子使得其晶体结构有一定的破损;从图6(c)和图6(d)可以看出两种不同催化剂催化反应后其晶体表面均沉积有一定量的颗粒状积炭。

图6 反应前、后催化剂的SEM表征Fig.6 SEM images of fresh and spent catalyst samples
2.2.2 TGA表征 由图7可知,两种不同的反应后催化剂均有两个不同的失重区间40~200℃,480~680℃。40~200℃范围内的失重,是样品吸附的水分在受热的条件下被释放而引起的失重,另外一个失重区间内的失重是由于催化剂积炭被燃烧引起的质量损失[4]。
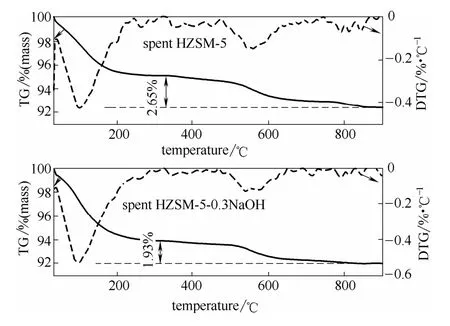
图7 反应后催化剂的失重曲线与失重速率曲线Fig.7 TG and DTG curves of catalyst samples
不考虑催化剂样品吸附的水分,改性HZSM-5热解木质素后催化剂的积炭量(1.93%)小于未改性HZSM-5热解木质素后催化剂的积炭量(2.65%)。改性 HZSM-5晶体内的晶体内新引入一部分介孔[13,23-26],与未改性HZSM-5相比,当木质素热解产物在新产生的介孔内扩散时,其扩散路径相对降低,因此木质素二次裂解产物在催化剂孔道内发生再聚合反应生成积炭的机会相应减少[25,27-28],所以改性后催化剂参与催化热解反应时其积炭量会相对降低。
2.2.3 FTIR表征 为了分析催化剂积炭的性质[29],选取反应后催化剂FTIR曲线的两个波数段(2800~3000 cm-1和1300~1800 cm-1)进行去卷积洛伦兹拟合[30-31],结果见图8、图9。2800~3000 cm-1范围内的峰代表脂肪族CHx的(对称和非对称)伸缩振动峰以及单环芳香类的伸缩振动峰;1300~1800 cm-1范围内的峰代表聚缩芳烃类、共轭烯烃类以及一些脂肪族类化合物的弯曲振动峰[30,32]。
波数在2955 cm-1处的峰为终端—CH3基团的C—H伸缩振动峰;2925 cm-1峰为—CH2或—CH基团的C—H伸缩振动峰;2895 cm-1峰为—CH2或—CH基团中C—H伸缩振动峰;2870 cm-1峰为终端—CH3基团的C—H伸缩振动峰;2855 cm-1峰为—CH2基团的C—H伸缩振动峰[30]。1710 cm-1峰代表着不饱和的羰基结构;1625 cm-1峰代表了芳烃的共轭结构,1590 cm-1峰代表了积炭分子组分中的缩聚芳香环结构;1430 cm-1峰代表了链烷烃中的C—H弯曲振动;1365 cm-1峰是由于富碳类化合物中双键的伸缩振动[29-31,33]。
图10是选取图 8和图 9两个特征波数范围2800~3000 cm-1和1300~1700 cm-1内所拟合出的洛伦兹峰进行峰面积归一化处理,计算出各峰的峰面积百分比所得。
从图 10(a)中可以看出对于两种不同催化剂催化反应后,催化剂积炭中的—CH2或—CH(2925 cm-1)的相对含量几乎为终端—CH3(2955 cm-1)相对含量的4倍,这说明催化剂积炭中含有长链的脂肪烃或者其中含有大量的环烷烃结构或者不饱和烃结构。从图10(a)中可以看出,改性HZSM-5积炭中的—CH2或—CH(2925 cm-1)的相对含量要小于未改性HZSM-5积炭中的相对含量。
从图10(b)中可以看出,两种催化积炭中具有共轭结构的芳烃类化合物(1625 cm-1)占据了绝大部分,改性HZSM-5积炭中的芳烃结构(1625 cm-1)的相对含量要高于未改性HZSM-5积炭中的相对含量,表明改性催化剂催化热解木质素过程中发生了更多的芳构化反应。图10(b)进一步证明了改性后催化剂积炭中链烃(1430 cm-1)的相对含量低于未改性催化剂积炭链烃中的相对含量,同时不饱和双键(1365 cm-1)的相对含量低于未改性催化剂积炭中的相对含量。对于两种不同的反应后催化剂,导致催化剂失活的主要积炭为1590 cm-1峰所对应的缩聚芳烃结构[30,33],从图10中可以看出对于这两种不同催化剂的1590 cm-1峰的相对含量区别不是特别明显,该峰在改性后催化剂积炭中的相对含量稍低于未改性的相对含量。
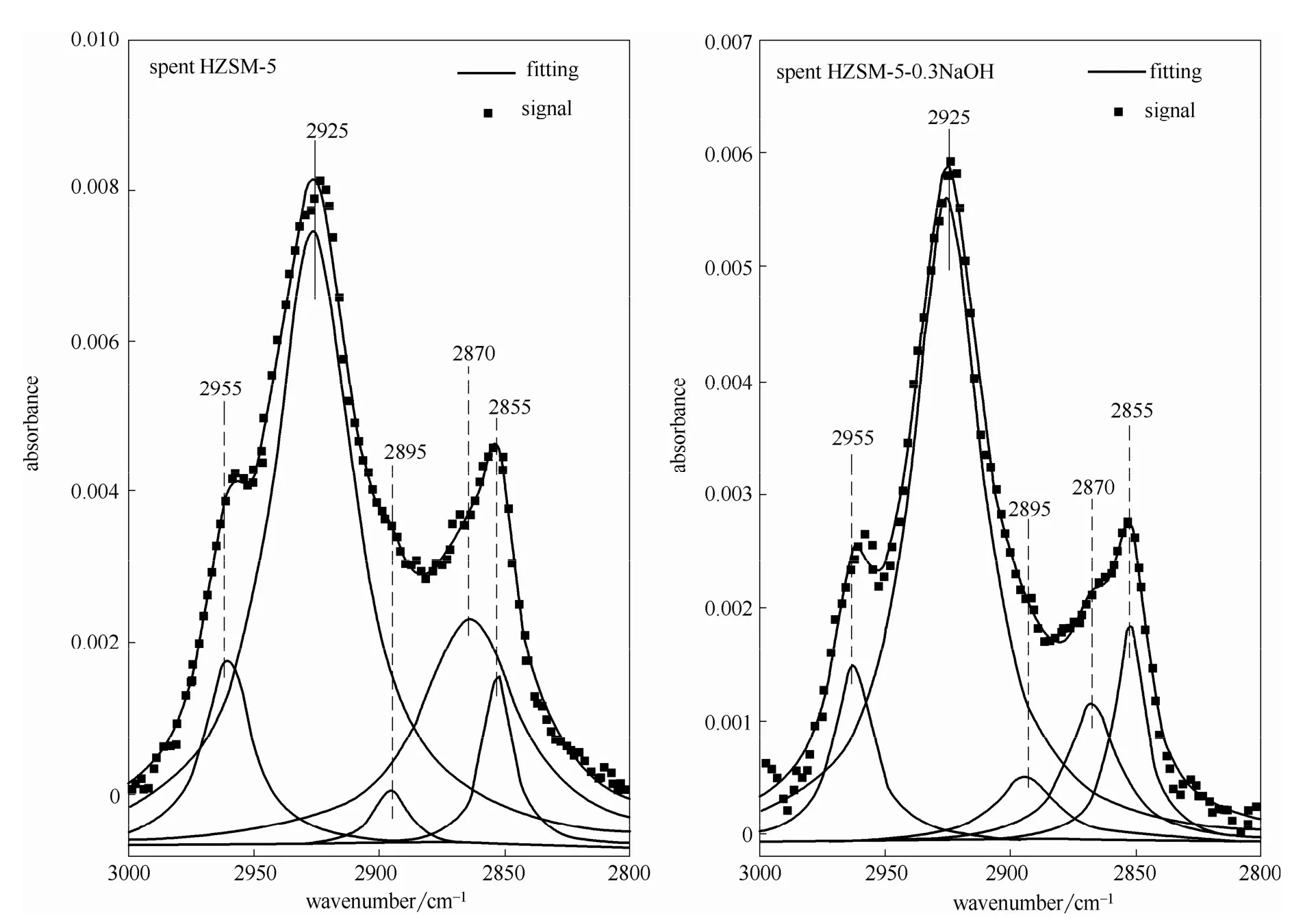
图8 反应后催化剂的红外光谱波数范围在2800~3000 cm-1的去卷积分峰拟合图Fig.8 Deconvolution fitting of vibrational bands in region 2800—3000 cm-1of spent catalyst samples
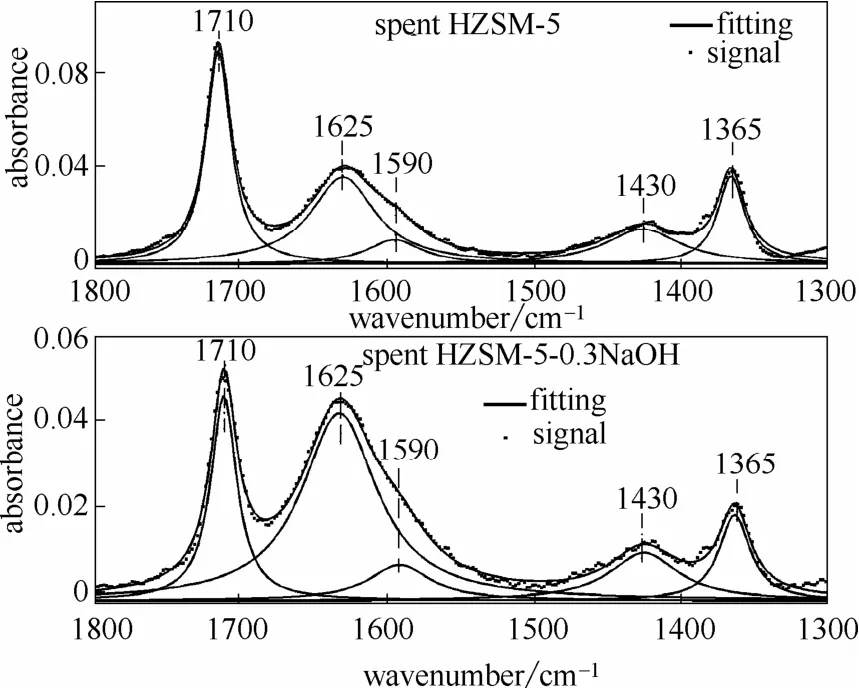
图9 反应后催化剂的红外光谱波数范围在1300~1800 cm-1的去卷积分峰拟合图Fig.9 Deconvolution fitting of vibrational bands in region 1300—1800 cm-1 of spent catalyst samples
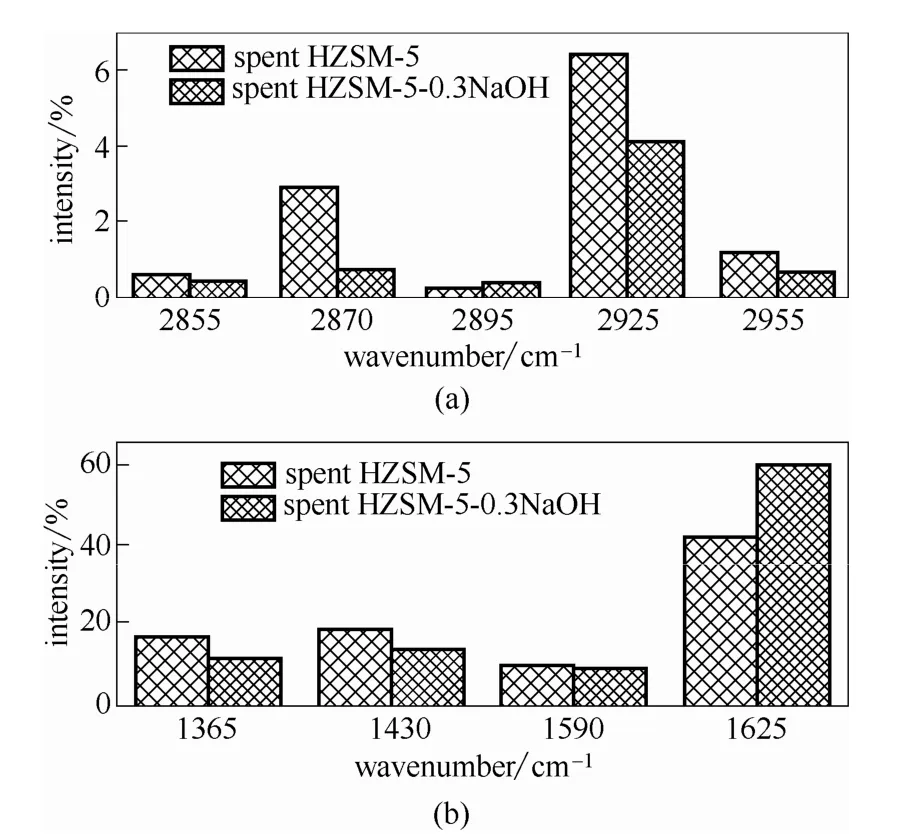
图10 波数范围在2800~3000 cm-1和1300~1700 cm-1内的拟合峰的相对强度Fig.10 Fraction of intensities of characteristic vibrational bands between 2800—3000 cm-1 and 1300—1700 cm-1
综上可以看出改性后催化剂积炭中的脂肪烃相对含量小于未改性催化剂的相对含量,芳烃的相对含量高于未改性催化剂积炭的相对含量。这是由于HZSM-5催化剂具有良好的孔道择形催化效果,改性后催化剂的孔径尺寸有所增加,这样为木质素催化热解生成的脂肪链烃在催化剂的活性位点上发生进一步环化、芳构化等反应生成聚合度较高的积炭大分子等提供足够的空间,因此其积炭组分中芳烃类结构的含量相对较多。
2.2.4 UV-Vis表征 紫外光谱(UV-Vis)法是一种鉴别催化剂积炭的不饱和性以及芳香性的方法[29-30,34]。
图11中,240~290 nm处的吸收峰代表了覆盖在催化剂表面的非均相积炭结构[30],310 nm峰为单烯基碳正离子的特征吸收峰,380 nm峰为二烯基碳正离子的特征吸收峰[35],410 nm峰代表六甲基取代苯离子或者五甲基取代苯离子结构,480 nm峰代表含有碳正离子聚合的甲基取代萘环结构,580 nm处的峰则表示具有更多碳正离子聚合的高聚结构[32]。对样品的紫外吸收光谱图进行去卷积处理(峰的解叠),拟合出了若干洛伦兹峰[30],拟合的各吸收峰的峰位置以及各峰的相对含量(峰面积百分比)列于表2。
从表2中可以看出未改性HZSM-5积炭的紫外可见吸收峰在380 nm以前的相对含量要高于改性后催化剂积炭的相对含量,改性后HZSM-5积炭的紫外可见吸收峰在410 nm以后的相对含量要高于未改性的相对含量,这表明高聚合度化合物在改性后催化剂积炭中的含量相对较高。
在木质素催化热解过程中,催化剂积炭主要是由于木质素热解生成的积炭前体在催化剂的酸性位点上发生再聚合反应而生成的。对于改性后的催化剂,其孔径增加,可接近的酸性位点的数量也随之增加,积炭前体接触到催化剂的酸性位点的机会也相应增加,同时由于改性后催化剂的孔径相对增大,这为孔内积炭进一步聚合生成高聚合度的积炭分子提供了一定的生长空间。因此改性后催化剂积炭中高聚合类化合物的相对含量要低于未改性催化剂积炭的相对含量。
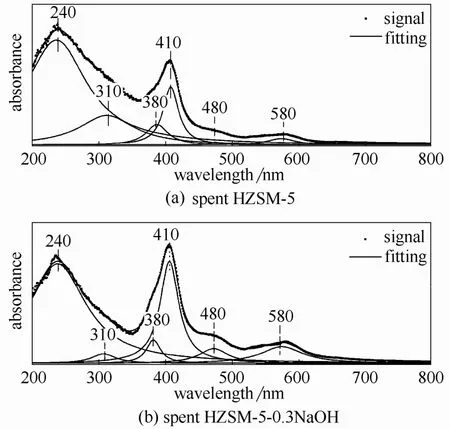
图11 反应后催化剂的紫外可见吸收光谱表征Fig.11 UV-Vis of spent catalyst samples
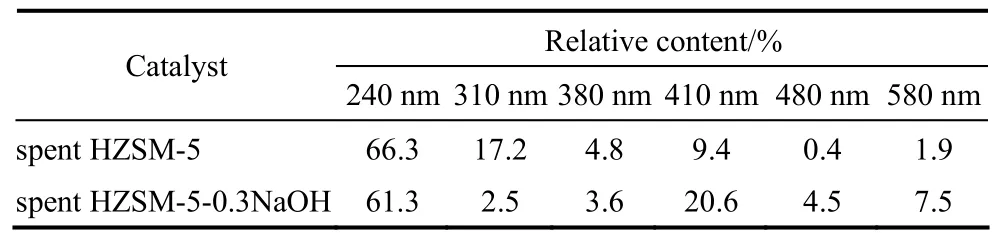
表2 反应后催化剂紫外谱图中各拟合峰相对含量Table 2 Characteristic fraction of intensities of peaks assigned in UV-Vis of spent catalysts
3 结 论
0.3 mol·L-1NaOH改性HZSM-5分子筛和未改性HZSM-5分子筛催化热解木质素的活性均随着实验循环次数的增加而逐渐降低。改性分子筛在改性时其骨架结构遭到破坏,经再生后其活性恢复能力要弱于未改性催化剂。对于反应后的两种催化剂,积炭堵塞孔道以及催化剂活性位点中毒均为其失活的原因,催化剂积炭可以通过高温煅烧的方式除去,但其活性位点中毒很难再恢复。由于积炭前体在改性HZSM-5分子筛内的扩散路径减小,积炭前体能容易地从孔内扩散出来,因此改性后催化剂的积炭生成量要低于未改性催化剂,且其催化剂生成积炭组分中聚合芳烃的含量相对较多。
[1]ZHANG M,RESENDE F L P,MOUTSOGLOU A.Catalytic fast pyrolysis of aspen ligninviaPy-GC/MS[J].Fuel,2014,116:358-369.
[2]MULLEN C A,BOATENG A A.Catalytic pyrolysis-GC/MS of lignin from several sources[J].Fuel Processing Technology,2010,91(11):1446-1458.
[3]LI X Y,SU L,WANG Y J,et al.Catalytic fast pyrolysis of Kraft lignin with HZSM-5 zeolite for producing aromatic hydrocarbons[J].Frontiers of Environmental Science & Engineering,2012,6(3):295-303.
[4]MA Z Q,TROUSSARD E,VAN BOKHOVEN J A.Controlling the selectivity to chemicals from ligninviacatalytic fast pyrolysis[J].Applied Catalysis A:General,2012,423/424:130-136.
[5]LI C,ZHAO X,WANG A,et al.Catalytic transformation of lignin for the production of chemicals and fuels[J].Chemical Reviews,2015,115(21):11559-11624.
[6]WANG K,KIM K H,BROWN R C.Catalytic pyrolysis of individual components of lignocellulosic biomass[J].Green Chemistry,2014,16(2):727-735.
[7]REZAEI P S,SHAFAGHAT H,DAUD W M A W.Production of green aromatics and olefins by catalytic cracking of oxygenate compounds derived from biomass pyrolysis:a review[J].Applied Catalysis A:General,2014,469:490-511.
[8]ZHOU G,JENSEN P A,LE D M,et al.Direct upgrading of fast pyrolysis lignin vapor over the HZSM-5 catalyst[J].Green Chemistry,2016,18(7):1965-1975.
[9]YU Y Q,LI X Y,SU L,et al.The role of shape selectivity in catalytic fast pyrolysis of lignin with zeolite catalysts[J].Applied Catalysis A:General,2012,447/448:115-123.
[10]JACKSON M A,COMPTON D L,BOATENG A A.Screening heterogeneous catalysts for the pyrolysis of lignin[J].Journal of Analytical and Applied Pyrolysis,2009,85(1/2):226-230.
[11]JAE J,TOMPSETT G A,FOSTER A J,et al.Investigation into the shape selectivity of zeolite catalysts for biomass conversion[J].Journal of Catalysis,2011,279(2):257-268.
[12]PARK H J,HEO H S,JEON J K,et al.Highly valuable chemicals production from catalytic upgrading of radiata pine sawdust-derived pyrolytic vapors over mesoporous MFI zeolites[J].Applied Catalysis B:Environmental,2010,95(3/4):365-373.
[13]LI J,LI X,ZHOU G,et al.Catalytic fast pyrolysis of biomass with mesoporous ZSM-5 zeolites prepared by desilication with NaOH solutions[J].Applied Catalysis A:General,2014,470:115-122.
[14]DU S,GAMLIEL D P,GIOTTO M V,et al.Coke formation of model compounds relevant to pyrolysis bio-oil over ZSM-5[J].Applied Catalysis A:General,2016,513:67-81.
[15]尹海云,李小华,张蓉仙,等.HZSM-5在线提质生物油及催化剂失活机理分析[J].燃料化学学报,2014,42(9):1077-1086.YIN H Y,LI X H,ZHANG R X,et al.Online catalytic cracking of bio-oil over HZSM-5 zeolite and analysis of catalyst deactivation[J].Journal of Fuel Chemistry and Technology,2014,42(9):1077-1086.
[16]GAYUBO A G,AGUAYO A T,ATUTXA A,et al.Deactivation of a HZSM-5 zeolite catalyst in the transformation of the aqueous fraction of biomass pyrolysis oil into hydrocarbons[J].Energy & Fuels,2004,18(6):792-797.
[17]GAYUBO A G,AGUAYO A T,ATUTXA A,et al.Transformation of oxygenate components of biomass pyrolysis oil on a HZSM-5 zeolite(Ⅰ ):Alcohols and phenols[J].Industrial & Engineering Chemistry Research,2004,43(11):2619-2626.
[18]JUN Y,LEE S,LEE K,et al.Effects of secondary mesoporosity and zeolite crystallinity on catalyst deactivation of ZSM-5 in propanal conversion[J].Microporous and Mesoporous Materials,2017,245:16-23.
[19]DING K,ZHONG Z,WANG J,et al.Effects of alkali-treated hierarchical HZSM-5 zeolites on the production of aromatic hydrocarbons from catalytic fast pyrolysis of waste cardboard[J].Journal of Analytical and Applied Pyrolysis,2017,125:153-161.
[20]SAMOLADA M C,PAPAFOTICA A,VASALOS I A.Catalyst evaluation for catalytic biomass pyrolysis[J].Energy & Fuels,2000,14(6):1161-1167.
[21]GAYUBO A G,VALLE B,AGUAYO A T,et al.Pyrolytic lignin removal for the valorization of biomass pyrolysis crude bio-oil by catalytic transformation[J].Journal of Chemical Technology &Biotechnology,2010,85(1):132-144.
[22]MA Z Q,VAN BOKHOVEN J A.Deactivation and regeneration of H-USY zeolite during lignin catalytic fast pyrolysis[J].ChemCatChem,2012,4(12):2036-2044.
[23]HOFF T C,GARDNER D W,THILAKARATNE R,et al.Elucidating the effect of desilication on aluminum-rich ZSM-5 zeolite and its consequences on biomass catalytic fast pyrolysis[J].Applied Catalysis A:General,2017,529:68-78.
[24]SADOWSKA K,WACH A,OLEJNICZAK Z,et al.Hierarchic zeolites:zeolite ZSM-5 desilicated with NaOH and NaOH/tetrabutylamine hydroxide[J].Microporous and Mesoporous Materials,2013,167:82-88.
[25]ZHU X,LOBBAN L L,MALLINSON R G,et al.Tailoring the mesopore structure of HZSM-5 to control product distribution in the conversion of propanal[J].Journal of Catalysis,2010,271(1):88-98.
[26]XIN H,LI X,FANG Y,et al.Catalytic dehydration of ethanol over post-treated ZSM-5 zeolites[J].Journal of Catalysis,2014,312:204-215.
[27]PÉREZ-RAMÍREZ J,ABELLÓ S,BONILLA A,et al.Tailored mesoporosity development in zeolite crystals by partial detemplation and desilication[J].Advanced Functional Materials,2009,19(1):164-172.
[28]MEI C,WEN P,LIU Z,et al.Selective production of propylene from methanol:Mesoporosity development in high silica HZSM-5[J].Journal of Catalysis,2008,258(1):243-249.
[29]BAUER F,KARGE H G.Characterization of Coke on Zeolites[M].Springer Berlin Heidelberg,2006:264-284.
[30]CASTA O P,ELORDI G,OLAZAR M,et al.Insights into the coke deposited on HZSM-5,Hβ and HY zeolites during the cracking of polyethylene[J].Applied Catalysis B:Environmental,2011,104(1/2):91-100.
[31]LI Y,ZHANG C,LIU Y,et al.Coke formation on the surface of Ni/HZSM-5 and Ni-Cu/HZSM-5 catalysts during bio-oil hydrodeoxygenation[J].Fuel,2017,189:23-31.
[32]PALUMBO L,BONINO F,BEATO P,et al.Conversion of methanol to hydrocarbons:spectroscopic characterization of carbonaceous species formed over H-ZSM-5[J].The Journal of Physical Chemistry C,2008,112(26):9710-9716.
[33]PARK J W,SEO G.IR study on methanol-to-olefin reaction over zeolites with different pore structures and acidities[J].Applied Catalysis A:General,2009,356(2):180-188.
[34]HUNGER M.Applications ofin situspectroscopy in zeolite catalysis[J].Microporous and Mesoporous Materials,2005,82(3):241-255.
[35]KIRICSI I,F RSTER H,TASI G,et al.Generation,characterization,and transformations of unsaturated carbenium ions in zeolites[J].Chemical Reviews,1999,99(8):2085-2114.
date:2017-06-13.
Dr.ZHANG Changsen,zhancs@zzu.edu.cn
supported by the International Cooperation Science Foundation of Henan Province (172102410041).
Deactivation analysis of catalyst for modified HZSM-5 catalytic lignin pyrolysis
TANG Songshan1,3,PAN Zeyou1,2,ZHANG Changsen1,2,WANG Dengtai1,2,XUE Xiangfei1,2,CAO Yunfeng3,LIU Yonggang1,2,ZHANG Ruiqin1,2
(1College of Chemistry and Molecular Engineering,Zhengzhou University,Zhengzhou450001,Henan,China;2Environmental Chemistry & Low Carbon Technologies Key Laboratory of Henan Province,Zhengzhou450001,Henan,China;3Topfond Pharmaceutical Co.,Ltd.,Zhumadian463000,Henan,China)
The circulation and regeneration experiments were employed to evaluate the lifetime of the two different catalysts (HZSM-5 and 0.3 mol·L-1NaOH modified HZSM-5).The spent and regenerated catalysts were characterized by N2adsorption-desorption and NH3-TPD techniques to analyse the reason of deactivation,and the spent catalysts were characterized by SEM,TGA,FTIR and UV-Vis techniques to analyse the coke deposited on the catalyst.The deactivation of the two different zeolites was become more and more serious along with the increase of the number of cycles.The difference was that the performance of 0.3 mol·L-1NaOH modified HZSM-5 was better than the one of unmodified HZSM-5 after four cycles.The coke of the spent catalysts was removed through calcination in air at high temperature and the catalytic activity was recovered to a large extent.The performance of 0.3 mol·L-1NaOH modified HZSM-5 was still better than the one of unmodified HZSM-5 after regeneration.The amount of coke deposited on the modified HZSM-5 was less than the one of unmodified HZSM-5.The coke of the modified HZSM-5 contains more condensed aromatics relatively.
HZSM-5 zeolites; catalytic pyrolysis; lignin; deactivation; regeneration; coke
TQ 032.4
A
0438—1157(2017)12—4739—11
10.11949/j.issn.0438-1157.20170764
2017-06-13收到初稿,2017-07-18收到修改稿。
联系人:张长森。
唐松山(1991—),男,硕士研究生。
河南省国际合作基金项目(172102410041)。
猜你喜欢
内燃机与动力装置(2022年1期)2022-03-21
湖北农机化(2020年4期)2020-07-24
石油炼制与化工(2020年7期)2020-01-05
上海包装(2019年8期)2019-11-11
汽车维护与修理(2018年7期)2018-10-13
筑路机械与施工机械化(2017年5期)2017-08-31
天津造纸(2016年1期)2017-01-15
天津大学学报(自然科学与工程技术版)(2015年10期)2015-12-29
中国造纸学报(2015年1期)2015-12-16
中国塑料(2014年4期)2014-10-17
