
表型正常假性甲状旁腺功能减退症一例并文献复习
2017-12-19 05:45:39魏伟平方团育全会标陈道雄陈开宁
海南医学 2017年22期
魏伟平,方团育,全会标,陈道雄,陈开宁
(海南省人民医院内分泌科,海南 海口 570311)
表型正常假性甲状旁腺功能减退症一例并文献复习
魏伟平,方团育,全会标,陈道雄,陈开宁
(海南省人民医院内分泌科,海南 海口 570311)
假性甲状旁腺功能减退症;表型正常;文献复习
假性甲状旁腺功能减退症(pseudohypoparathyroidism,PHP)是一组罕见的由于外周靶器官对甲状旁腺激素(parathyroid hormone,PTH)抵抗而导致的遗传性疾病,临床表现多样,共同特征是具有甲旁减的生化改变(低血钙、高血磷),但由于靶组织对PTH无反应,表现为PTH正常或升高,多数PHP患者有脸圆、身材矮粗、短指(趾)畸形(掌骨和跖骨短小)等Albright遗传性骨营养不良症(albright hereditary osteodystrophy,AHO)畸形表现,而表型正常的少见,本文介绍我科收治的一例表型正常的PHP患者,以提高临床医师对该病的认识,现报道如下:
1 病例简介
患者,男,19岁,学生,因“反复肢体抽搐、僵硬5年”于2017年2月13日收入海南省人民医院内分泌科。患者5年前开始无明显诱因出现肢体抽搐、僵硬,以右侧肢体明显,无头晕、乏力,无肌强直,症状间断出现,伴有肢端麻木,可自行缓解,未治疗,近2年发作次数较前减少。2个月余前患者活动时跌倒,伴四肢抽搐,无口吐白沫,无意识不清,在当地医院查头颅CT提示脑内广泛性钙化,为进一步诊治至我院门诊,检查提示:血钾 3.19 mmol/L、血钙 1.73 mmol/L(2.03~2.54 mmol/L),甲状旁腺激素测定(发光法):全型甲状旁腺素 236.60 pg/mL(参考值15~68.3 pg/mL),头颅CT(见图1):双侧小脑齿状核及双侧大脑半球多发对称性钙化灶,性质待定,甲状旁腺功能低下?血管病变?建议MRI检查,为进一步诊治收收入我科。起病后,患者睡眠差、多梦、无精神异常。患者既往体健,无食物及药物过敏史,无家族史。体格检查:生命体征平稳,发育正常,体型正常,智力正常,神志清楚,毛发分布正常,无全身浅表淋巴结未触及肿大。突眼征阴性,颈软,无抵抗,甲状腺未扪及肿大。心肺腹无异常,无趾指畸形,四肢肌力、肌张力正常。病理反射阴性。面神经叩击征阴性,束臂加压试验阳性。入院后完善相关辅助检查:血、粪便常规、尿常规、肝、肾功能正常,电解质:血钙 1.67 mmol/L(参考值2.03~2.54 mmol/L)、血磷 2.01 mmol/L(参考值0.9~1.34 mmol/L),血镁、血钾、碱性磷酸酶正常,骨碱性磷酸酶为240 IU/L(正常值不超过200 IU/L,临界值200~250 IU/L)25-羟维生素D 26.4 ng/mL(参考值25~150 ng/mL),全型甲状旁腺素231.10 pg/mL(参考值15~68.3 pg/mL),性腺六项正常,皮质醇 330 nmol/L(08:00),皮质醇<22.07 nmol/L(00:00),尿酸化功能:pH值6.72,促肾上腺皮质激素31.80 pg/mL(08:00),促肾上腺皮质激素8.86 pg/mL(00:00),血气分析:酸碱度7.37、剩余碱-2.80 mmol/L,24 h尿磷11.77 mmol/L,24 h尿肌酐 9590.0 μmol/L,24 h尿钙0.41 mmol/L,肾小管磷重新收率:97.24%(参考值85%~95%)。骨显像示:全身骨显像未见典型恶性肿瘤骨转移征象,建议定期复查。甲状旁腺显像示:未见甲状旁腺功能亢进影像改变。泌尿系彩超示:前列腺钙化斑。双肾、输尿管未见异常声像。双肾血供丰富。腹部、心脏彩超正常。心电图、脑电图正常。胸部+骨盆+腰椎+胸椎DR片示:胸椎未见异常。S1腰化、隐裂。考虑L5/S1椎间盘病变,建议进一步检查。骨盆、双髋关节未见异常。心肺未见异常。手正斜位片:双手骨质未见明显异常。头颅MRI:双侧丘脑、双侧基底节辐射冠区对称性异常信号,结合CT,考虑Fahr综合征或代谢性疾病(甲旁亢?)请结合临床实验室检查。广州金域假性甲旁减1a型GNAS基因测序结果:阴性。结合病史及实验室检查,后考虑诊断为:假性甲状旁腺机能减退症,给予10%葡萄糖酸钙10 mL静脉注射,口服碳酸钙1.5 g tid,骨化三醇胶丸0.25µg qd治疗,后症状改善出院,随访半年无抽搐发作。
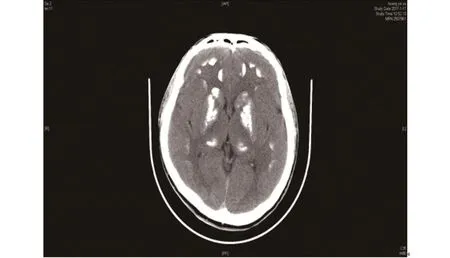
图1 头颅CT提示双侧小脑齿状核及双侧大脑半球多发对称性钙化灶
2 讨 论
PHP是极少见的遗传性甲状旁腺疾病,表型正常的更为罕见,该病为常染色体显性或隐性疾病,与编码G蛋白α亚单位(Gsa)的基因GNAS失活性突变导致靶器官(骨和肾)对PTH不反应有关,女性多见,发病率为男性的2倍,多为母系突发传递发病,有家族聚集性,也可散发,2岁后出现症状多见,一般10岁后较明显,20岁以后发病者罕见,发病年龄平均8~9岁[1]。
PHP的病因为PTH受体或受体后缺陷,以至靶细胞(骨、肾)对PTH无反应或敏感性降低所致。甲状旁腺分泌的PTH与靶器官细胞上的受体结合,激活与受体耦联的鸟苷酸结合蛋白(G蛋白),进一步激活腺苷酸环化酶(AC),AC在镁离子的帮助下,将ATP转化为cAMP,cAMP作为第二信使进一步激活蛋白激酶A,导致蛋白磷酸化,产生生物学效应,上述过程中任一个环节缺陷,都会造成靶器官对PTH抵抗,导致PHP,故一般认为PTH对靶器官的作用由cAMP(腺苷酸环化酶)调节,与鸟苷酸结合蛋白(G蛋白)有关[2]。
根据靶细胞对PTH的反应性不同,PHP可以分为两种类型[3-4]:(1)Ⅰ型,由于靶器官细胞膜上PTH受体发生缺陷,不能与PTH结合,或虽结合但也不能激活腺苷酸环化酶(AC),不能形成cAMP,故外源性PTH刺激后,检测尿cAMP和尿磷均无变化,而正常人尿cAMP可在注射后30 min升高50~100倍,尿磷排出量也可明显增加。根据患者症状、体征、分子缺陷等因素,PHPI型可进一步分为Ⅰa、Ⅰb和IⅠc三个亚型,其中常见的是PHPⅠa型,又称为Albright遗传性骨营养不良症,是PHP的主要类型,其发病机制是由于PTH的受体蛋白即GS蛋白a亚基活性下降,功能缺陷所致,该型患者通常有典型AHO的特殊体型(即AHO畸形),血细胞Gsa活性降低,由于甲状腺激素和生长激素等均由G蛋白介导,Gsa活性下降可导致多发性内分泌缺陷,如性腺功能减退、促甲状腺素(TSH)抵抗、生长激素缺乏等,部分患者伴有智力低下。该型的分子病因为GNAS1基因突变,GNAS1基因主要编码Gsa蛋白质,是一种印记基因,由12个内含子和13个外显子组成,通过不同的启动子和共同的2~13号外显子转录、翻译产生多种基因产物[5],近年来研究提示PHPⅠa型多有GNAS1基因1~13号外显子失活性突变[6]。Ⅰb型表现为对PTH的抵抗,其GSa蛋白活性通常是正常的,通常无AHO畸形,部分患者骨组织可对PTH仍有反应,骨吸收常可增加.严重时可导致骨质疏松及纤维性骨炎,常见无多发性内分泌缺陷,多为散发,也有家族性,发病机制尚不清楚,有文献报道是因为GNASl基因启动子区3个差异甲基化区域(DMR)的异常甲基化所致[7-8]。ⅠC型极少见,其Gsa活性正常,具有AHO畸形和多发激素抵抗,分子机制不明,可能与腺苷酸环化酶的催化单位异常有关,目前认为可能与GNAS1基因无关[2]。(2)PHPⅡ型存在受体后缺陷,靶细胞对cAMP无反应,导致PTH与受体结合后无法发挥其生理效应,故外源性PTH刺激后,检测尿cAMP升高,但尿磷无变化,可用于Ⅰ型和Ⅱ型PHP的鉴别。一般认为PHPⅡ型无AHO畸形,仅表现为PTH抵抗,不具有明确的遗传和家族基础,可能为AHO的一种亚型,部分患者有PTH受体多态性[2]。另外还存在一种特殊类型即假一假性甲状旁腺功能减退症(PPHP),存在AHO畸形,但不伴PTH抵抗和多发内分泌缺陷,血生化正常,其分子机制为GSa活性下降或GNAS1突变[3-4]。
PHP多为儿童时期起病,临床表现主要与PTH抵抗导致的低钙、高磷血症有关[9],血钙水平轻度降低时,可出现感觉异常,表现为口周或肢端麻木和刺痛,常不引起注意,当血钙降低到一定程度时,表现为典型的手足抽搐(血清钙一般<2 mmol/L)症状,甚至出现癫痫样发作,导致意识丧失、昏迷等,临床上常为患者就诊的首发及主要症状,易误诊为特发性癫痫,因此,对于以癫痫样发作临床表现者,接诊医师除常规行脑电图、CT等检查外,还应行血钙磷检测,若出现血钙降低、血磷升高,则需进一步完善PTH等检查。病情较轻者,一般不自行发作手足抽搐,可通过面神经叩击征试验(Chvostek征),束臂加压试验(Trousseau征)诱发。有些轻症或久病患者主要表现为头痛、焦虑、烦躁等精神症状,有时可误诊为癔症,需注意鉴别。长期低血钙导致白内障、晶状体浑浊亦常见,严重时影响视力。除上述甲状旁腺功能减退的症状之外,PHP大多伴有发育异常、智力发育障碍及AHO躯体畸形(圆脸、颈短、身材粗矮、短趾、短指等)。PHP的生化特点表现为低血钙、高血磷,低尿钙、尿磷,计算肾小管磷的重吸收率明显升高,由于靶器官对PTH不反应,故低血钙反馈刺激甲状旁腺过度合成分泌PTH,故血PTH不降低反而升高,这也是该病与甲状旁腺功能减退症的主要鉴别依据。碱性磷酸酶(ALP)通常正常,但Ⅰb型患者ALP可升高,考虑与该型患者骨组织对升高的PTH仍有反应有关[10]。其他检查中,心电图常有QT间期延长、T波低平等非特异性改变,X线骨骼照片可见第4第5掌(趾)骨较粗短、锁骨增宽、前臂骨弯曲等异常表现。长期高磷可导致脑内、软组织等处钙离子沉积,出现转移性钙化,故头颅CT可见颅内钙化,尤其是基底核区,钙化特点为双侧、对称、多发,出现较早,可能是癫痫的重要原因,也是其特征性表现,故部分患者脑电图也可有损害表现。本病例患者有PTH抵抗的临床表现和生化异常,如反复发生肢体僵硬、手足抽搐、高血钙、低血磷、肾小管磷的重吸收率明显升高,而血PTH浓度升高,同时颅脑CT检查提示双侧小脑齿状核及双侧大脑半球多发对称性钙化灶,可诊断为PHP,可进一步行PTH兴奋试验对其进行分型,即给予外源性PTH,检测尿cAMP、尿磷,若尿cAMP增加,尿磷不增加,则考虑为Ⅱ型;尿cAMP、尿磷均不增加,则考虑为Ⅰ型。由于实验室无法检测尿cAMP,故无法进行PTH兴奋试验,从已有检查结果及病史来看,由于患者无典型的AHO体征,基因检测已排除Ⅰa型,故分型上考虑为Ⅰb型或Ⅱ型可能性大,结合患者骨ALP升高,提示其骨组织对PTH仍有反应,考虑Ⅰb型的可能性更大,若有条件行PTH兴奋试验、GSa活性检测或GNAS1基因检测,即可进一步明确分型。
PHP目前尚不能根治,其治疗类似于特发性甲旁减,主要目标是控制病情,缓解症状,急性低血钙时,患者表现为手足抽搐,喉痉挛,甚至癫痫大发作,需要静脉补充钙剂,可使用10%葡萄糖酸钙10~20 mL静脉推注,1~2 h后重复给药,若症状难以缓解,可考虑持续静脉滴注治疗,10%葡萄糖酸钙溶液100 mL用生理盐水或葡萄糖液稀释后使用,速度为每小时不超过元素钙4 mg/kg体重,同时密切监测血钙,避免高血钙发生,3周内服用洋地黄制剂患者慎用[10]。慢性低血钙患者主要采用维生索D及钙剂,使血钙接近正常、血磷下降,防止四肢抽搐与异位钙化,血生化指标纠正后,PTH代偿性分泌增加也可得到纠正,只是维生素D及钙剂使用量通常比特发性甲旁减低,由于个体反应的差异性,必须确定每个患者的最佳治疗方案,以维持正常的血钙值及尿钙,但因尿磷排泄障碍,应避免过多奶制品、肉、豆制品等高磷食物的摄入,避免导致高磷血症加重异位钙化。另外由于1a羟化酶的作用依赖于PTH,当PTH抵抗时,维生素D只能转化为25-(OH)D3,因此,使用骨化三醇治疗的效果要更优于维生素D,对于低钙血症需终生治疗,临床上应早期诊断、及时治疗,以防止不可逆的病理损害,PPHP无需特殊治疗,只需随访血钙变化即可[10]。
[1]Weinstein LS,Liu J,Sakamoto A,et al.Minireview:GNAS,normal and abnormalfunctions[J].JEndocrinology,2004,145(12):5459-5464.
[2]姚佳琦,任跃忠.假性甲状旁腺功能减退症的研究进展[J].国际内分泌代谢杂志,2007,27(5):321-324.
[3]Mantovani G,Spada A.Mutations in the Gs alpha gene causing hormone resistance[J].Best Pract Res Clin Endocrinol Metab,2006,20(4):501-513.
[4]Jan dBS,Ding C,Germain-Lee E,et al.Discordance between genetic and epigenetic defects in pseudohypoparathyroidism type lb revealed by inconsistent loss of maternal imprinting at GNAS1[J].Am J Hum Genet,2003,73(2):314-322.
[5]廖二元.G蛋白与内分泌代谢疾病[J].国际内分泌代谢杂志,2003,23(2):86-89.
[6]Thiele S,Wemer R,Ahrens W,et al.A disruptive mutationin exon 3 of the GNAS gene with albright hereditary osteodystrophy,normoealcemic pseudohypoparathyroidism,and selective long transcript variant Gsalpha-L deficiency[J].J Chin Endocrinol Metab,2007,92(5):1764-1768.
[7]Liu J,Nealon JG,Weinstein LS.Distinct patterns of abnormal GNAS Imprinting in familial and sporadic pseudohypoparathymidism type IB[J].Hum Mol Genet,2005,14(1):95-102.
[8]张小丽,孙良阁,刘爽,等.假性甲状旁腺功能减退症Ib型与GNAS1基因启动子甲基化的研究[J].中华内分泌代谢杂志,2010,26(11):973-975.
[9]密红翠,瞻台林芳,冯清仪,等.假性甲状旁腺机能减退症1例报告[J].中国医师杂志,2005,7(5):696.
[10]尹潍.代谢性骨病学[M].北京:人民卫生出版社,1989:342-355.
R582+.2
D
1003—6350(2017)22—3769—03
10.3969/j.issn.1003-6350.2017.22.054
魏伟平。E-mail:410233661@qq.com
2017-06-05)
猜你喜欢
中国临床医学影像杂志(2022年6期)2022-07-26 07:17:26
中国民间疗法(2021年13期)2021-08-30 08:56:48
成都医学院学报(2021年2期)2021-07-19 08:35:20
保健与生活(2020年13期)2020-07-24 19:44:38
西南国防医药(2016年7期)2016-12-01 06:01:26
西南军医(2016年3期)2016-01-23 02:17:49
微创泌尿外科杂志(2015年4期)2015-12-23 11:18:22
中国医学影像学杂志(2015年9期)2015-12-15 11:03:28
家庭医学·下半月(2015年5期)2015-05-30 10:48:04
山西卫生健康职业学院学报(2014年5期)2014-04-09 06:36:48
