
Caroli综合征合并常染色体显性遗传性多囊肾1例报告
2017-12-16 02:40:43陈洋溢袁继丽刘成海
临床肝胆病杂志 2017年12期
陈洋溢, 袁继丽, 邢 枫, 刘成海,2,3
(1 上海中医药大学附属曙光医院 肝硬化科, 上海 201203; 2 上海市中医临床重点实验室,上海 201203; 3 上海高校中医内科学E-研究院, 上海 201203)
Caroli综合征合并常染色体显性遗传性多囊肾1例报告
陈洋溢1, 袁继丽1, 邢 枫1, 刘成海1,2,3
(1 上海中医药大学附属曙光医院 肝硬化科, 上海 201203; 2 上海市中医临床重点实验室,上海 201203; 3 上海高校中医内科学E-研究院, 上海 201203)
Caroli病; 多囊肾疾病; 病例报告
1 病例资料
患者女性,8岁,于2014年6月2日突发“呕血、色红,量约200 ml”就诊于南京某医院。既往史、个人史无特殊。实验室检查:血常规:WBC 6.43×109/L,RBC 3.74×1012/L,PLT 61×109/L,Hb 86 g/L;肝功能:ALT 29 IU/L,AST 41 IU/L,AKP 140 U/L,TBil 14.5 μmol/L,Alb 49 g/L。乙型肝炎HbsAg阴性,HBV DNA低于检测下限,抗HCV阴性;HAV、HEV阴性。胃镜示“食管下段静脉曲张,红色征阳性”(图1),当时予内镜下食管曲张静脉硬化剂注射治疗。CT提示考虑Caroli病 Ⅱ 型(图2)。经对症治疗后出院。2014年10月在外院诊治,磁共振胰胆管造影(MRCP)示“肝大,脾大,肝内胆管扩张,肝总管炎性狭窄,双肾囊肿”(图3)。2014年12月基因检测显示PKD-1杂合突变(表1)。今为求进一步治疗,于2015年12月就诊于曙光医院。查体:慢性肝病面容,皮肤巩膜无黄染,蜘蛛痣及肝掌阴性。颈静脉无怒张,肝颈静脉回流征阴性。心肺查体未见异常。腹软,无腹壁静脉曲张,全腹无压痛,移动性浊音阴性。肝右肋下未及,脾左肋下5 cm,质硬,表面光滑,无触痛。双下肢无水肿。诊断为Caroli综合征伴常染色体显性遗传多囊肾疾病脾功能亢进。患者已进展至肝硬化失代偿期,建议尽早行肝移植术。给予保肝治疗,并纠正低血小板血症。经治疗后患者症情稳定。
2 讨论
Caroli病是一种常染色隐性遗传疾病,全称为交通性海绵状肝内胆管囊状扩张症。由法国学者Caroli于1958年首先报道并描述,其发病率约为1/100万[1]。该病可见于任何年龄段,男性发病人数多于女性。其发病隐匿,症状复杂,病变可以累及肝的一段、一个局部或双侧肝内胆管。主要病理变化为肝内胆管呈多发性囊状扩张。临床上以有无肝纤维化和门静脉高压将该病分为两型。Ⅰ型为单纯肝内胆管扩张,肝小叶结构正常,无肝纤维化或门静脉高压,常可伴胆囊结石和胆管炎。Ⅱ型为肝内胆管扩张伴肝纤维化、门静脉高压,肝小叶结构被破坏。其中Ⅰ型还可以根据扩张的位置分为Ⅰa和Ⅰb型。前者囊状扩张在肝的周围局限于一侧或一叶,后者囊状扩张位于肝的中央与肝门部主要的肝内胆管相通。Caroli病的发病机制是胆管板畸形导致胆管系统发育异常[2]。其起病隐匿,特别是Ⅰ型患者,可无任何明显不适,仅有轻微生化学指标异常。常发现时就已是Ⅱ型,甚至是肝硬化失代偿期,故临床上Ⅰ型较Ⅱ型更少见[3]。目前Caroli病的诊断依据主要为影像学证据,包括CT、MRCP、超声成像、经内镜逆行胰胆管造影,尤其以CT、MRCP最为重要。其能显示囊状影与肝内其他小胆管之间的关系,可作为诊断此病的重要依据。影像学典型征像有:“蝌蚪征”(囊状扩张的胆管与细小的胆管相通,形似蝌蚪);“悬挂征”(囊状影犹如悬挂在胆道树上的果实一般);“中心圆点征”(增强CT上可见低密度的囊状影中间有一强化的点状血管影,其实质为扩张的胆管包绕肝内血管)[4-5]。该病病理结果主要表现为肝内胆管囊状扩张,纤维增生,成纤维细胞增生反应,周围有淋巴细胞等炎症细胞浸润,胆管上皮增生,甚至可见异型增生或癌变。文献[6]报道癌变率为7%,从发现该病到发生癌变,一般时长为3.3年。Caroli病无特异性的治疗方法。外科手术是唯一的确定性治疗方式。Ⅰ型可行根治性手术,晚期可行肝移植术。本文患儿已进展至肝硬化失代偿期,故建议尽早行肝移植手术。Starzl移植中心的Caroli病患者在术后随访中,1、5、10年的存活率分别高达76%、65%和56%[7]。

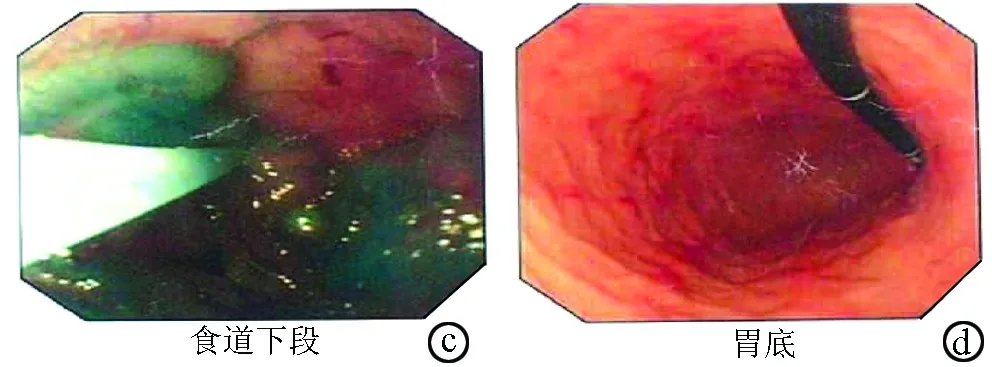
图1胃镜:食管静脉曲张3级a:食道中段见4条青蓝色蚓状隆起; b,c:食道下段及贲门曲张静脉迂曲增粗呈串珠样,表面见红色征;d:胃底黏膜红相为主,未见明显扩张
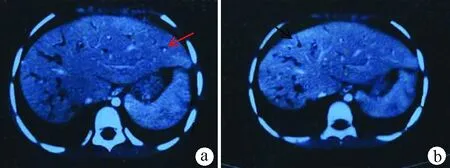
图2增强CT a:肝脏密度增高部分呈结节状表现,肝裂增宽,其内见点状高密度影(中心圆点征,红箭头);b:肝内胆管广泛扩张; 囊状扩张与柱状小胆管相通(蝌蚪征,黑箭头),增强后肝内囊性灶未见强化。脾脏形态增大明显
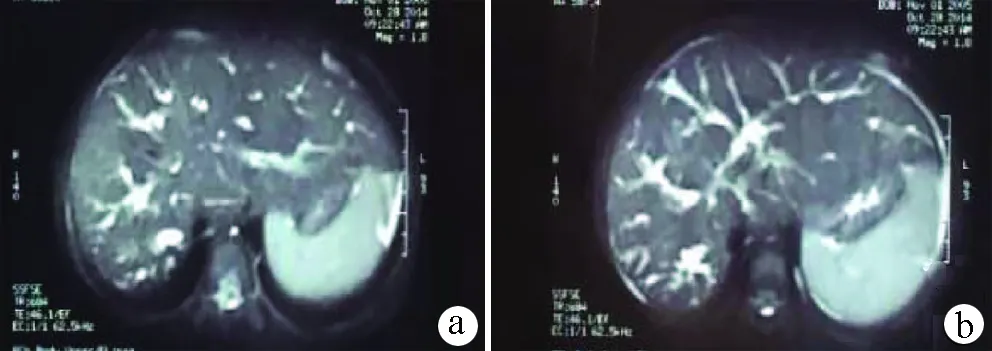
图3 MRCP a:肝脏形态饱满,肝右叶萎缩,肝左叶及尾状叶增大,肝内胆管囊状扩张;b:肝内胆管树显示基本清晰,肝内胆管扩张,远端胆管扩张明显,肝总管管腔狭窄;脾脏增大明显

注:het,杂合子;AR,隐性遗传;AD,显性遗传 该患者存在多位点的突变,其中PKD1基因的突变可发病。
大量临床证据[8]表明该病与遗传性多囊肾疾病呈高度相关,目前已明确多囊肾是由于PKD1、PKD2、PKHD1这三个基因位点突变引起的,其中PKD1、PKD2突变导致常染色体显性遗传性多囊肾疾病,PKHD1突变导致常染色体隐性遗传多囊肾疾病。上述3种基因分别编码PC-1、PC-2和fibrocystin三种蛋白,此三种蛋白主要定位于肾小管与胆管上皮中的原纤毛上,而这些原纤毛被认为有着重要的生理病理功能,由上述三种蛋白缺失而导致的原纤毛功能障碍极有可能是肝肾囊性病变的主要病因[9]。Caroli病合并常染色体显性遗传多囊肾疾病既往也有报道,其症状多变,可表现为反复的胆管炎症、胆道感染以及肝内胆管扩张[10-13],但该病主要还是与常染色体隐性遗传性多囊肾病和肾消耗病相伴随[14-17],合并常染色体显性遗传多囊肾疾病的案例较少见,而本文患儿已进展至肝硬化、门静脉高压,更为罕见。本病例提示:对于常染色体显性遗传多囊肾疾病患者,特别是反复发生胆管炎、胆道感染、胆结石而又无明确诱因的患者,需排除患者合并Caroli病可能。
[1] CAROLI J, SOUPAULT R, KOSSAKOWSKI J, et al. La dilatation polykystique congenital des voies biliaires intrahepatiques: essai declassification[J]. Sem Hop Paris, 1958, 34(2): 2128-2135.
[2] DESMET VJ. Congenital diseases of intrahepatic bile ducts: variations on the theme “ductal plate malformation”[J]. Hepatology, 1992, 16(4): 1069-1083.
[3] SHEN XP, SHEN BX, ZHAO QZ, et al. Diagnosis of Caroli disease by CT and MRI[J]. J Chin Physician, 2005, 8(7): 1045-1047. (in Chinese)
沈新平, 沈比先, 赵清洲, 等. Caroli病的CT和MRI诊断[J]. 中国医师杂志, 2005, 8(7): 1045-1047.
[4] LU N, GUO WL, CHEN LY, et al. CT and MRI diagnosis of Caroli disease(analysis of 8 cases)[J]. J Chin Clin Med Imaging, 2006, 11(17): 656-658. (in Chinese)
陆娜, 郭文力, 陈丽英, 等. Caroli病的影像诊断(附8例分析)[J]. 中国临床医学影像杂志, 2006, 11(17): 656-658.
[5] PERRICONE G, VANZULLI A. Education and imaging.Hepatology:" central dot sign" of Caroli syndrome[J].J Gastroenterol Hepatol, 2015, 30(2): 234.
[6] HUANG ZQ, LIU YX, ZHOU NX. Problems in the surgical treatment of Caroli’s disease[J]. Chin J Surg, 1995, 11(33): 666-668. (in Chinese)
黄志强, 刘永雄, 周宁新. Caroli病外科治疗中的问题[J]. 中华外科杂志, 1995, 11(33): 666-668.
[7] HABIB S, SHAKIL O, COUTO OF, et al. Caroli’s disease and orthotopic liver transplantation[J]. Liver Transpl, 2006, 12(3): 416-421.
[8] SENYUZ OF, YESILDAG E, KURUOGLU S, et al. Caroli’s disease in children: Is it commonly misdiagnosed?[J]. Acta Pediatrica, 2005, 94(1): 117-120.
[9] MASYUK AI, MASYUK TV, SPLINTER PL, et al. Cholangiocyte cilia detect changes in luminal fluid flow and transmit them into intracellular Ca2+and cAMP signaling[J]. Gastroenterology, 2006, 131(3): 911-920.
[10] JORDON D, HARPAZ N, THUNG S. Caroli’s disease and adult polycystic kidney disease: a rarely recognized association[J]. Liver, 1989, 9(1): 30-35.
[11] DRANSSART M, COGNET F, MOUSSON C, et al. MR cholangiography in the evaluation of hepatic and biliary abnormalities in autosomal dominant polycystic kidney disease:study of 93 patients[J]. J Comput Assist Tomogr, 2002, 26(2): 237-242.
[12] ISHIKAWA I, CHIKAMOTO E, NAKAMURA M, et al. High incidence of common bile duct dilatation in autosomal dominant polycystic kidney disease patients[J]. Am J Kidney Dis, 1996, 27(3): 321-326.
[13] TERADA T, NAKAMURA Y. Congenital biliary dilatation in autosomal dominant adult polycystic disease of the liver and kidneys[J]. Arch Pathol Lab Med, 1988, 112(11): 1113-1116.
[14] WEN J. Congenital hepatic fibrosis in autosomal recessive polycystic kidney disease[J]. Clin Transl Sci, 2011, 4(6): 460-465.
[15] LIANG JJ, KAMATH PS. Caroli syndrome[J]. Mayo Clin Proc, 2013, 88(6): e59.
[16] PARKE,LEE JM,AHN YH,et al.Hepatorenal fibrocystic diseases in children[J]. Pediatr Nephrol, 2016, 31(1): 113-119.
[17] OBUSEZ EC, UDAYASANKAR U. Autosomal recessive polycystic kidney disease with Caroli syndrome[J].J Urol, 2015, 193(2): 679-680.
Caroli’ssyndromewithautosomaldominantpolycystickidneydisease:acasereport
CHENYangyi,YUANJili,XINGFeng,etal.
(Departmentofcirrhosis,ShuguangHospitalAffiliatedtoShanghaiUniversityofTraditionalChineseMedicine,Shanghai201203,China)
caroli disease; polycystic kidney, autosomal recessive; case reports
R575
B
1001-5256(2017)12-2403-02
10.3969/j.issn.1001-5256.2017.12.032
2017-07-15;修回日期:2017-08-15。 作者简介:陈洋溢(1991-),男,主要从事中西医结合慢性肝病防治工作。 通信作者:刘成海,电子信箱:chenghailiu@hotmail.com。
引证本文:CHEN YY, YUAN JL, XING F, et al. Caroli’s syndrome with autosomal dominant polycystic kidney disease: a case report[J]. J Clin Hepatol, 2017, 33(12): 2403-2404. (in Chinese)
陈洋溢, 袁继丽, 邢枫, 等. Caroli综合征合并常染色体显性遗传性多囊肾1例报告[J]. 临床肝胆病杂志, 2017, 33(12): 2403-2404.
(本文编辑:林 姣)
猜你喜欢
中国现代医生(2022年19期)2022-11-04 10:13:29
昆明医科大学学报(2022年4期)2022-05-23 13:04:50
基层中医药(2021年8期)2021-11-02 06:24:54
江苏卫生保健(2021年9期)2021-03-27 16:25:04
家庭医药(2021年2期)2021-03-09 06:48:09
保健医苑(2020年6期)2020-12-04 01:33:11
消费导刊(2017年24期)2018-01-31 01:29:31
辽宁大学学报(哲学社会科学版)(2017年3期)2017-06-21 21:16:59
中学语文(2015年27期)2015-03-01 03:53:28
西南军医(2015年1期)2015-01-22 09:08:34
