
临床干血斑样本中12种抗凝血鼠药的液相色谱-串联质谱法同时定量检测
2017-11-29 08:15王怡君李嘉玮邱泽武谢剑炜
分析测试学报 2017年11期
王怡君,郭 磊,徐 斌,陈 佳,李嘉玮,邱泽武,谢剑炜*
(1.军事医学科学院毒物药物研究所,抗毒药物与毒理学国家重点实验室,北京 100850;2.军事医学科学院附属医院 中毒救治科,北京 100071)
临床干血斑样本中12种抗凝血鼠药的液相色谱-串联质谱法同时定量检测
王怡君1,郭 磊1,徐 斌1,陈 佳1,李嘉玮2,邱泽武2,谢剑炜1*
(1.军事医学科学院毒物药物研究所,抗毒药物与毒理学国家重点实验室,北京 100850;2.军事医学科学院附属医院 中毒救治科,北京 100071)
为应对临床中毒快检的需求,建立了干血斑样本中12种抗凝血鼠药的液相色谱-串联质谱定性定量方法。针对全打孔的干血斑样本,考察了滤纸卡种类、润湿步骤、提取溶剂种类对提取效果的影响。采用C18色谱柱进行分离,以乙酸铵(5 mmol/L)水溶液-乙酸铵(5 mmol/L)甲醇溶液为流动相进行梯度洗脱,在电喷雾负离子电离方式下使用三重四极多反应监测模式检测。结果表明,采用Whatman903滤纸卡为基底,以水为溶剂充分润湿,再以含内标的甲醇溶液提取5 min,各鼠药的提取率为66%~115%,且结果稳定(日内RSD小于15%)。除杀鼠酮的线性范围为20~500 ng/mL(r2=0.998 7)外,其它11种鼠药的线性范围为5~500 ng/mL(r2=0.998 8~0.999 6);12种鼠药的回收率为61%~105%,基质效应为71%~193%(日内RSD小于15%)。该方法准确、灵敏、快速且操作简便,成功应用于3例临床鼠药中毒病人样本的快速检测,为临床毒物检测和法医毒物分析的快速筛查和准确定量提供了新的技术方法。
干血斑;抗凝血杀鼠药;液相色谱-串联质谱法;临床中毒快检
因剧毒鼠药、农药等有毒化学品的误服或蓄意投毒造成的临床化学性中毒事件在我国每年均屡有发生,对民生造成了极大危害。临床化学性中毒救治亟需快速响应的毒物检测和临床诊断方法。临床干血斑技术的出现,满足了采样方便、损伤小、样本量低、便于运输及储存等需要,也便于临床中毒样品的监测及快速检测。干血斑是一种新兴的血液采集及保存方式,即将指尖或足尖血滴在滤纸卡上,常温下干燥2 h以上完成制备[1-2]。其采血量一般少于100 μL;操作简便,病人或其家属即可完成采血;蛋白质和酶以非活性方式存在于干血斑中,细菌的生长受到抑制,生物危害性低;不需低温储藏,运输时不需冷藏设备的辅助。目前,干血斑已应用于临床新生儿疾病筛查、药物代谢动力学研究及临床药物监测[3-4]等方面。例如,Heinig等[5]通过液质方法进行治疗药物的临床监测,发现干血斑样品与液态全血、液态血浆的药物分析数据一致、可靠。但到目前为止,干血斑多在疾病标志物、临床药物监测方面展开应用,对临床毒物监测的研究尚处于起步阶段[6-7]。
本文以常见的抗凝血鼠药化学性中毒为例,对临床干血斑样本的抗凝血鼠药检测进行方法学和初步应用研究。以香豆素类和茚二酮类为代表的第二代抗凝血杀鼠药,通过抑制环氧化物还原酶,阻止维生素K还原,降低维生素K依赖性凝血因子活性,导致各脏器及粘膜大面积出血,严重者可导致死亡[8]。第二代抗凝血鼠药具有脂溶性高、毒性强的特点,其半衰期长,在体内清除缓慢,病人治疗依从性差,常需长时间连续监测用药后体内清除进程[9-10]。目前,针对血样中抗凝血鼠药的分析检测方法主要包括液相色谱(LC)[11-12]、气相色谱-质谱(GC-MS)[13]及液相色谱-串联质谱技术(LC-MS/MS)[14-17]等。针对全血、组织等生物样本,需经有机溶剂提取、固相净化或富集等步骤进行前处理。本文在优化干血斑样本提取效率的基础上,建立了普适快速的前处理方法,继而基于液相色谱-三重四极杆串联质谱(LC-QqQ-MS/MS)技术,建立了12种常见抗凝血鼠药临床干血斑样本的定性定量检测方法,并成功应用于3例临床中毒样本的快速诊断,方法具有简便、准确快速、灵敏度高的特点。
1 实验部分
1.1 仪器、试剂与材料
Agilent 6430型高效液相色谱-三重四极杆串联质谱仪(LC-QqQ-MS/MS,Agilent公司,美国),配备电喷雾离子源;R200D电子天平(Sartorius,德国);M-S涡旋混合器(Scilogex公司,美国);1-14型高速离心机(Sigma公司,美国);Milli-QA10超纯水仪(Millipore公司,美国);Agilent ZORBAX Eclipse Plus C18色谱柱(2.1 mm×50 mm,1.8 μm)和保护柱(4.6 mm,0.2 μm,Agilent公司,美国);S-4800扫描电镜(Hitachi公司,日本)。
12种抗凝血鼠药对照品溶液(10 μg/mL),包括克灭鼠、华法林、杀鼠酮、杀鼠醚、氯灭鼠灵、敌鼠、氯敌鼠、溴敌隆、溴敌隆酮、鼠得克、氟鼠酮和大隆(纯度高于98%,Toronto Research Chemicals公司,加拿大)均由北京疾病预防控制中心配制并提供;霉酚酸(纯度高于98%,Ark Pharm公司,美国);甲醇和乙腈(色谱纯,Merck公司,德国);甲酸(纯度98%,百灵威公司,北京);乙酸铵(纯度高于98%,Acros Organics公司,美国);Milli-Q超纯水(电阻率为18.2 MΩ·cm)。
Whatman DMPK-A、DMPK-B、DMPK-C滤纸卡和903滤纸卡及打孔器(GE公司,美国)。离心管(2.0 mL,Axygen公司,美国);全血取自Wistar大鼠(许可证号:SCXK(京)2012-0001,饲养于军事医学科学院实验动物中心SPF级动物房);患者中毒血样由军事医学科学院附属医院中毒救治科提供。
1.2 溶液及样品制备
12种鼠药混合标准储备溶液:质量浓度为10 μg/mL,溶于甲醇,于-20 ℃保存。
12种鼠药混合标准工作溶液:取适量标准储备溶液,以甲醇逐级稀释至质量浓度为1 000、100 ng/mL,于4 ℃保存(需在1个月内使用完毕)。
质量控制(QC)溶液质量浓度分别为5、20、50、100、200、500 ng/mL。
内标储备溶液:称取2 mg霉酚酸,以甲醇配制成1 mg/mL溶液,于-20 ℃保存。
内标工作溶液:取适量内标储备溶液,以甲醇稀释至200 ng/mL,于4 ℃冰箱中保存(需在1个月内使用完毕)。
干血斑样本的制备:吸取20 μL全血滴在滤纸卡上(直径约1 cm),室温下干燥2 h即得。
1.3 样品前处理
选取整个干血斑,用打孔器取下,置于离心管中,加入20 μL水,待润湿后,加入80 μL内标溶液,涡旋5 min,移取全部溶液,以12 000 r/min离心5 min,吸取上层清液至进样瓶。
1.4 色谱条件
固定相:ZORBAX Eclipse Plus C18色谱柱(2.1 mm×50 mm,1.8 μm);保护柱(4.6 mm,0.2 μm);流动相:A为5 mmol/L 乙酸铵水溶液,B为5 mmol/L 乙酸铵的甲醇溶液;流速:0.2 mL/min;梯度洗脱程序:0~1.5 min,30%~80%B;1.5~6.0 min,80%~95%B;柱温:50 ℃;进样体积:5 μL。
1.5 质谱条件
离子源:电喷雾电离源(ESI源);扫描方式:负离子扫描;检测方式:多反应监测(MRM);喷雾压力40.0 psi,离子源温度100 ℃,雾化器温度:325 ℃;雾化器流量:10.0 L/min;毛细管电压:3.5 kV。
2 结果与讨论
所考察的12种抗凝血杀鼠药可分为香豆素类和茚二酮类两种,其油水分布系数(logD)介于1.6~9.3之间,即极性分布范围较宽,与滤纸卡间形成的作用力可能存在多种形式。按母核结构及logD值排序,将12 种鼠药分成4组:A组为杀鼠酮、敌鼠和氯敌鼠(茚二酮类,logD值1.6~5.3),B组为克灭鼠、华法林和氯灭鼠灵(香豆素类,logD值2.6~4.0),C组为杀鼠醚、溴敌隆和溴敌隆酮(logD值5.0~8.3),D组为鼠得克、氟鼠酮和大隆(logD值8.4~9.3),进行干血斑处理及提取条件等因素的考察。
2.1 滤纸卡类型
滤纸卡由于其自身材质和修饰方式的不同,与基质及待测化合物可能呈现出不同的相互作用,从而影响待测化合物的提取效率。对于20 μL全血,选用Whatman型滤纸卡(厚度0.50~0.74 mm),较之普通化学定量滤纸(厚度0.18 mm)更利于血液的集中分布,其扩散半径仅为0.5 cm。实验比较了903型、DMPK-A、-B、-C型4种类型的滤纸卡,其中903型和DMPK-C型未经任何化学修饰,DMPK-A型以自由基抑制剂、DMPK-B型以离液等成分进行修饰,均可起到变性蛋白、裂解细胞等作用[18]。各类型滤纸卡在滴加血液后的形态如图1所示。与其它3种滤纸卡相比,仅DMPK-A型卡上的干血斑出现明显的“咖啡环”效应,DMPK -B型卡上的干血斑呈现中央聚集,提示其上存在一定修饰。DMPK -C型卡和903卡上的血滴分布相对较为均匀。在扫描电镜下,DMPK-B型卡上纤维素表面更加粗糙。
针对12种抗凝血鼠药,以甲醇进行提取的结果表明,使用DMPK-A卡,未能检测到溴敌隆和溴敌隆酮,其它10种鼠药的绝对回收率在30%~98%之间。使用DMPK-B、DMPK-C卡、903卡,均可检测到12种鼠药,绝对回收率分别为10%~59%、43%~106%、65%~115%。4种类型的滤纸卡中,茚二酮类鼠药在DMPK-A、DMPK-C卡上得到了相对较高的回收率(60%~90%);而9种香豆素类抗凝血鼠药在903卡上的回收率最高(65%~115%)。大部分化合物在DMPK-C和 903卡上响应接近,考虑到903卡已获得FDA认证,且在临床上应用较为广泛,因此选用903滤纸卡进行后续研究。每组鼠药在相同滤纸卡上的响应差别小于10%,说明数据重复性较好,分组亦较为合适。
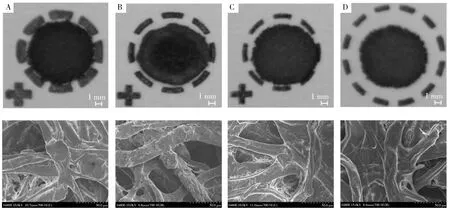
图1 光学显微镜(上)及扫描电镜(下)下的干血斑在各滤纸卡上的形态Fig.1 The pattern of dried blood spots on different kinds of filter paper cards under microscope(upper) and scanning electron microscope (lower)A:DMPK-A card,B:DMPK-B card,C:DMPK-C card,D:903 card
2.2 样品前处理步骤的优化
在干血斑中,滤纸不仅作为血液样本的承载基底,更能利用纤维素自身膨胀和吸附的特性与目标物质发生作用。当血液渗透滤纸时,经氢键作用,纤维素链间结合了大量水分子,其结构发生膨胀。同时,纤维素链相互交错形成空隙,小分子物质可进一步结合到纤维素链间,大分子物质则留于空隙中或置于空隙外。当水分丢失后,纤维素结构趋向恢复自然状态,各种物质分布于滤纸的表面或较深处[19]。因此若先加入适量水使滤纸膨胀后,有可能利于小分子物质从纤维素链间游离出来,从而提高提取效率,继而可再加入有机溶剂进行提取。结果表明,与直接用有机溶剂提取相比,先以水润湿再以有机溶剂提取的回收率提高了8%~30%。
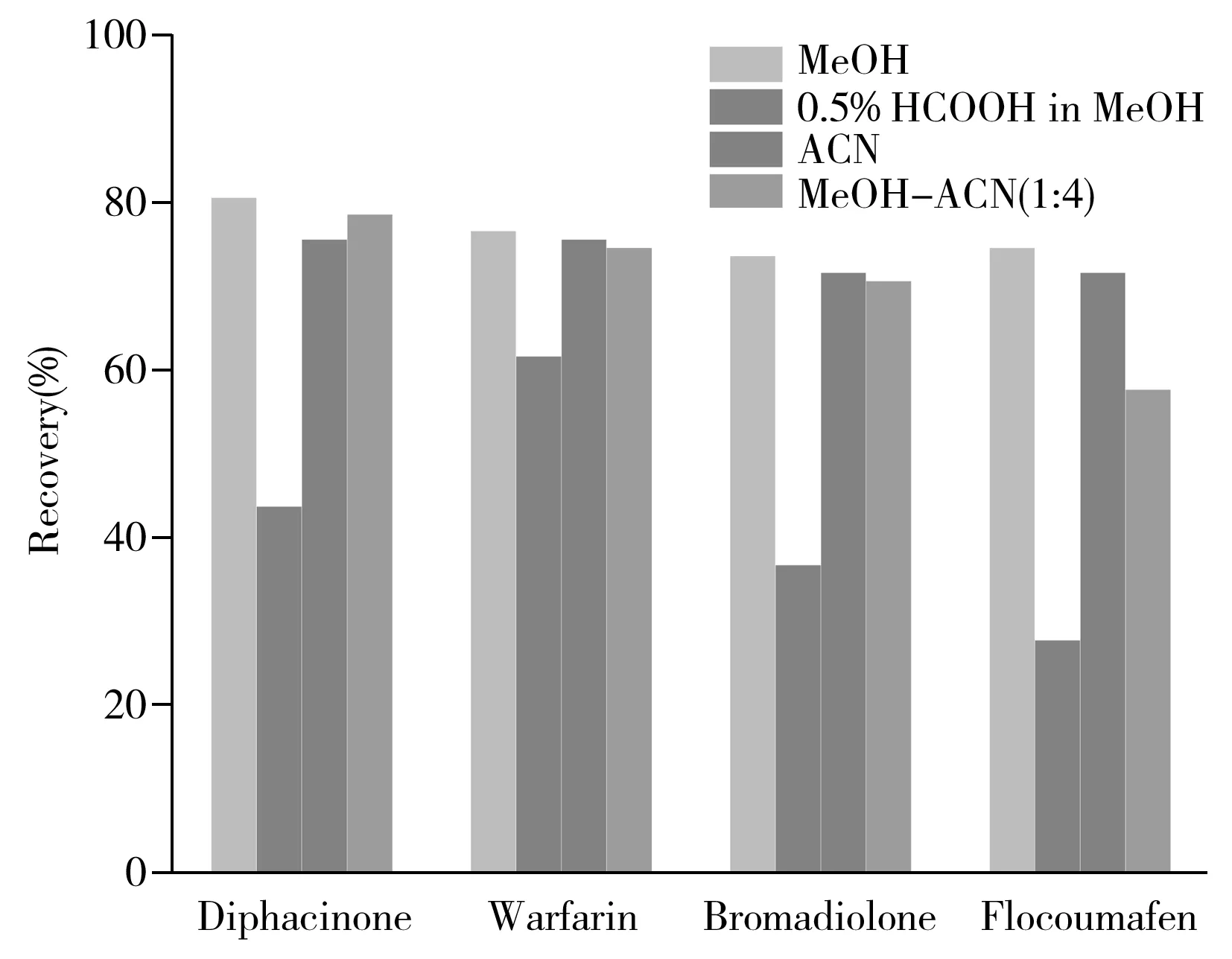
图2 4种溶剂的提取效果对比图Fig.2 The extraction efficiency by four kinds of solvents
提取溶剂种类方面,比较了甲醇、含0.5%甲酸的甲醇、乙腈及甲醇-乙腈(1∶4)的提取效果(图2),发现纯甲醇、乙腈提取时各鼠药的提取率较高(60%~115%),说明滤纸卡上抗凝血鼠药在提取溶剂中的分配与极性直接相关。因此本实验采用甲醇作为提取溶剂。
2.3 液相色谱条件优化
使用乙酸铵水溶液(A)-乙酸铵甲醇溶液(B)为流动相进行梯度洗脱,各化合物在B相为80%~95%的梯度洗脱范围内流出,各峰的保留时间与logD基本呈正相关。考察了CAPCELL CORE C18(2.1 mm×100 mm,2.7 μm)柱(Ⅰ)、Syncronis C18(2.1 mm×100 mm,1.7 μm)柱(Ⅱ)和ZORBAX Eclipse Plus C18(2.1 mm×50 mm,1.8 μm)柱(Ⅲ)的分离情况,结果显示,采用色谱柱Ⅰ时,华法林、杀鼠醚等峰形不佳,出现肩峰;采用色谱柱Ⅱ时,各化合物的峰形较好,但分离时间较长(达到14 min);采用色谱柱Ⅲ时,各化合物的峰形对称、分离好、洗脱时间短,故选用ZORBAX Eclipse Plus C18色谱柱。12种鼠药的MRM参数见表1,MRM色谱图见图3。
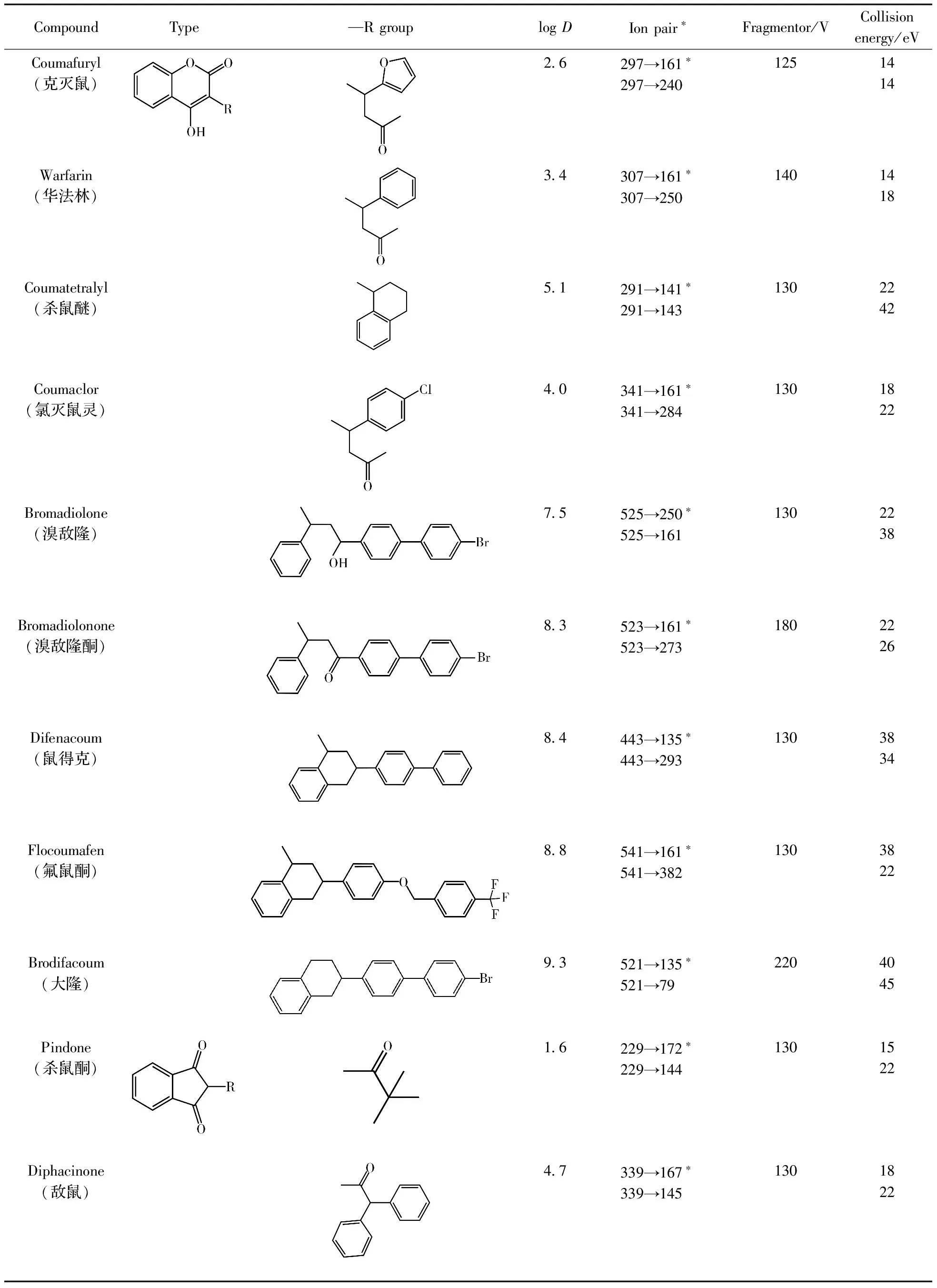
表1 12种抗凝血鼠药及内标的结构式、logD值及MRM参数Table 1 The structures,logD values and MRM parameters of 12 anticoagulant rodenticides
(续表1)
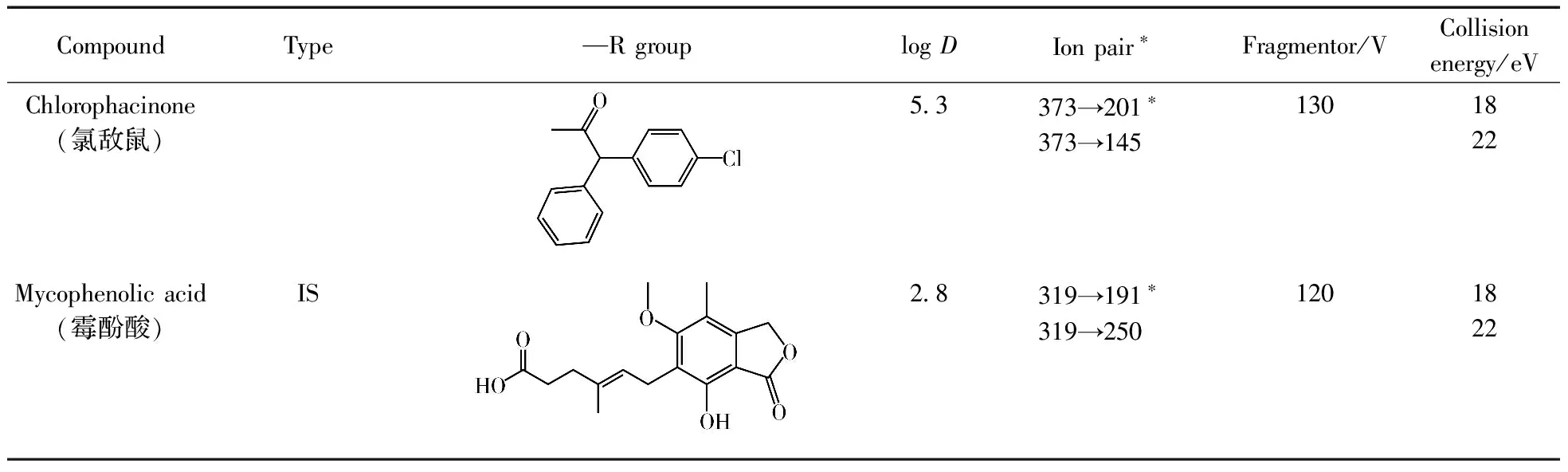
CompoundType—RgrouplogDIonpair∗Fragmentor/VCollisionenergy/eVChlorophacinone(氯敌鼠)5 3373→201∗373→1451301822Mycophenolicacid(霉酚酸)IS2 8319→191∗319→2501201822
*quantitative ion pair
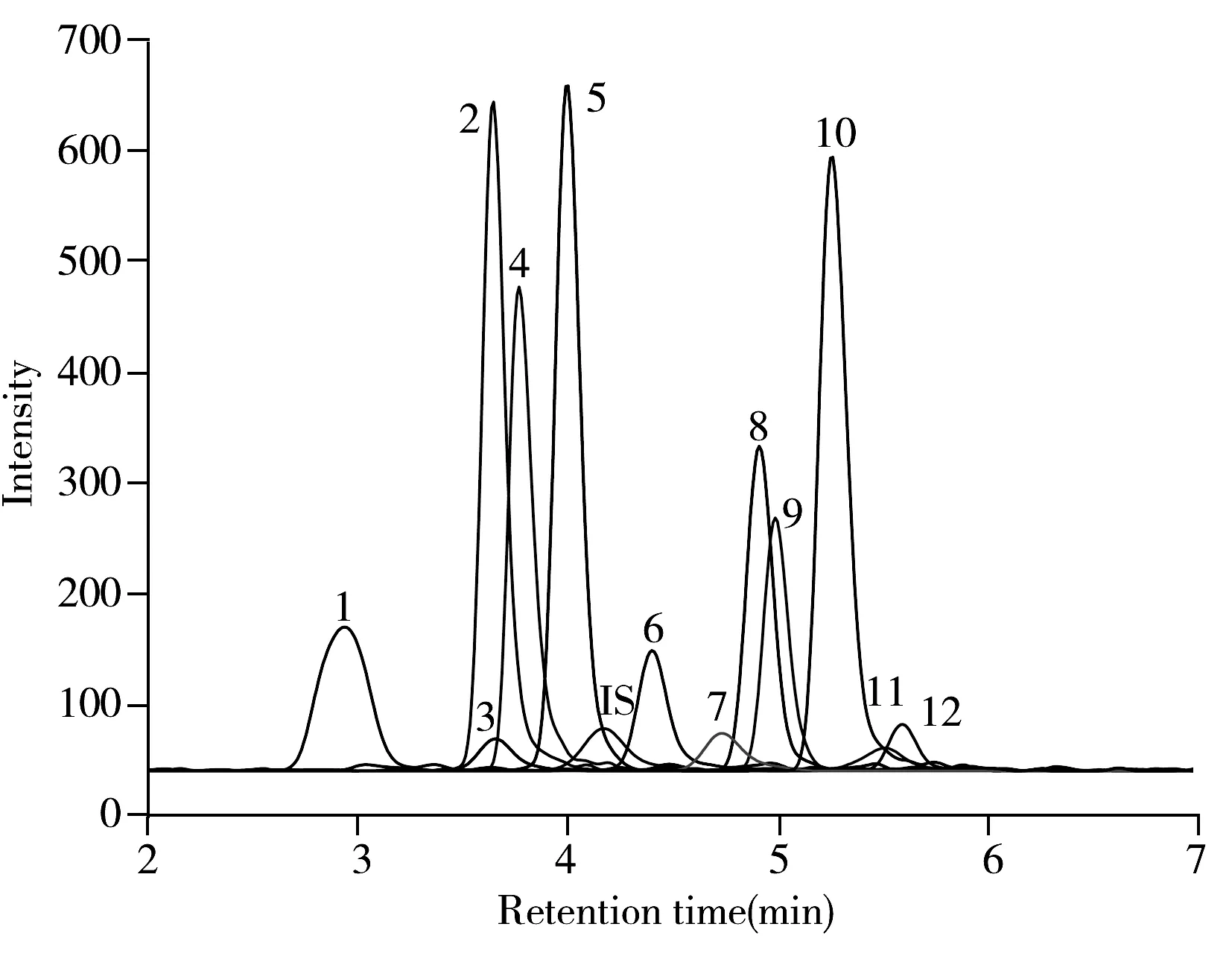
图3 12种抗凝血鼠药的MRM色谱图Fig.3 MRM chromatogram of 12 anticoagulant rodenticides peaks:1.coumafuryl;2.warfarin;3.pindone;4.coumatetralyl; 5.coumachlor;6.diphacinone;7.chlorophacinone; 8.bromadiolone;9.bromadiolonone;10.difenacoum; 11.flocoumafen;12.brodifacoum;IS:mycophenolic acid
2.4 方法学验证
2.4.1定量下限与标准曲线针对所制备的干血斑样品,以10倍信噪比(S/N=10)所对应的浓度作为定量下限(LOQ)。以化合物与内标的定量离子对峰面积比值(Y)为纵坐标,干血斑中化合物的质量浓度(X,ng/mL)为横坐标,以1/X加权拟合标准曲线。除杀鼠酮的线性范围为20~500 ng/mL(r2=0.998 7)外,其余11种鼠药的线性范围均为5~500 ng/mL(r2=0.998 8~0.999 6),满足方法学要求,结果见表2。
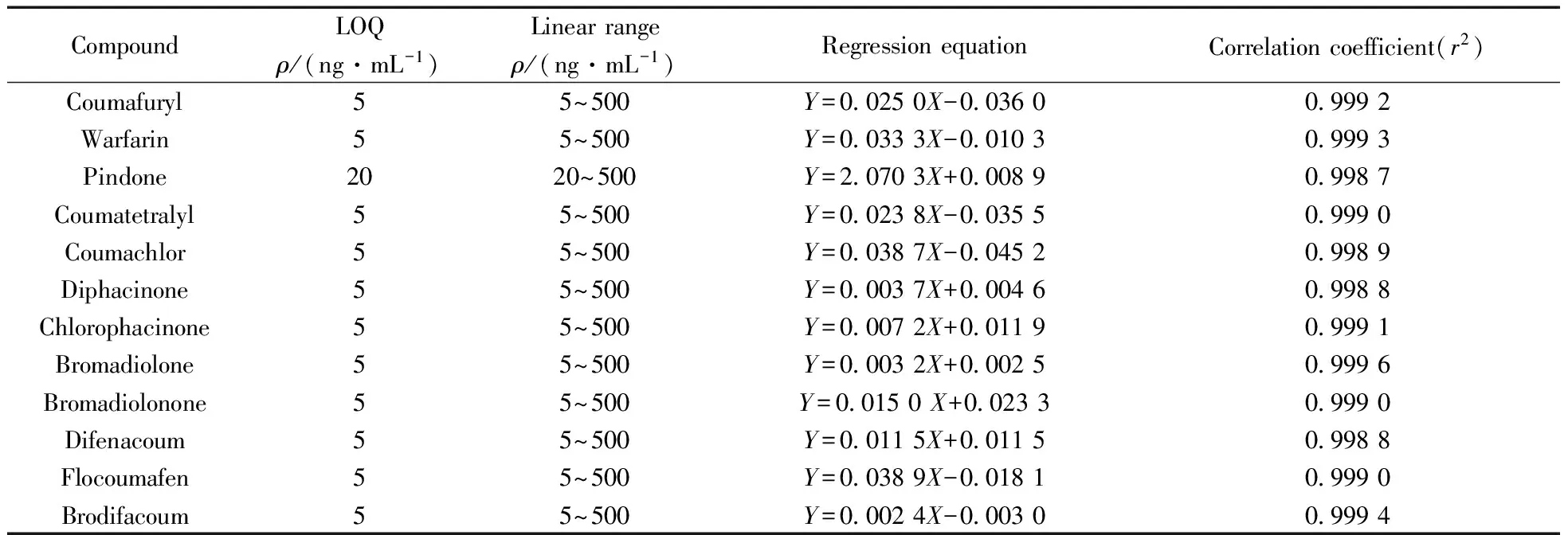
表2 12种鼠药的定量下限和线性范围Table 2 The LOQ and linear range of 12 anticoagulant rodenticides
2.4.2准确度与精密度对定量下限、低、中、高4个浓度进行考察,其中低浓度点为2倍定量下限,中浓度选取线性范围的中间浓度,高浓度点为80%定量上限。每个浓度独立制备5份干血斑样品,共测定3批样品,按照前处理方法操作,其精密度和准确度结果见表3。结果显示,各鼠药的批内和批间精密度、准确度均满足方法学要求。
2.4.3回收率与基质效应全血等复杂基质组分一般会通过影响被测物的离子化效率而影响待测物的响应,因此考察了基质效应。于低、高2个浓度分别制备3份样品,结果显示,该方法存在一定的基质增强作用,但其精密度尚能满足方法要求(RSD均小于15%,表3)。
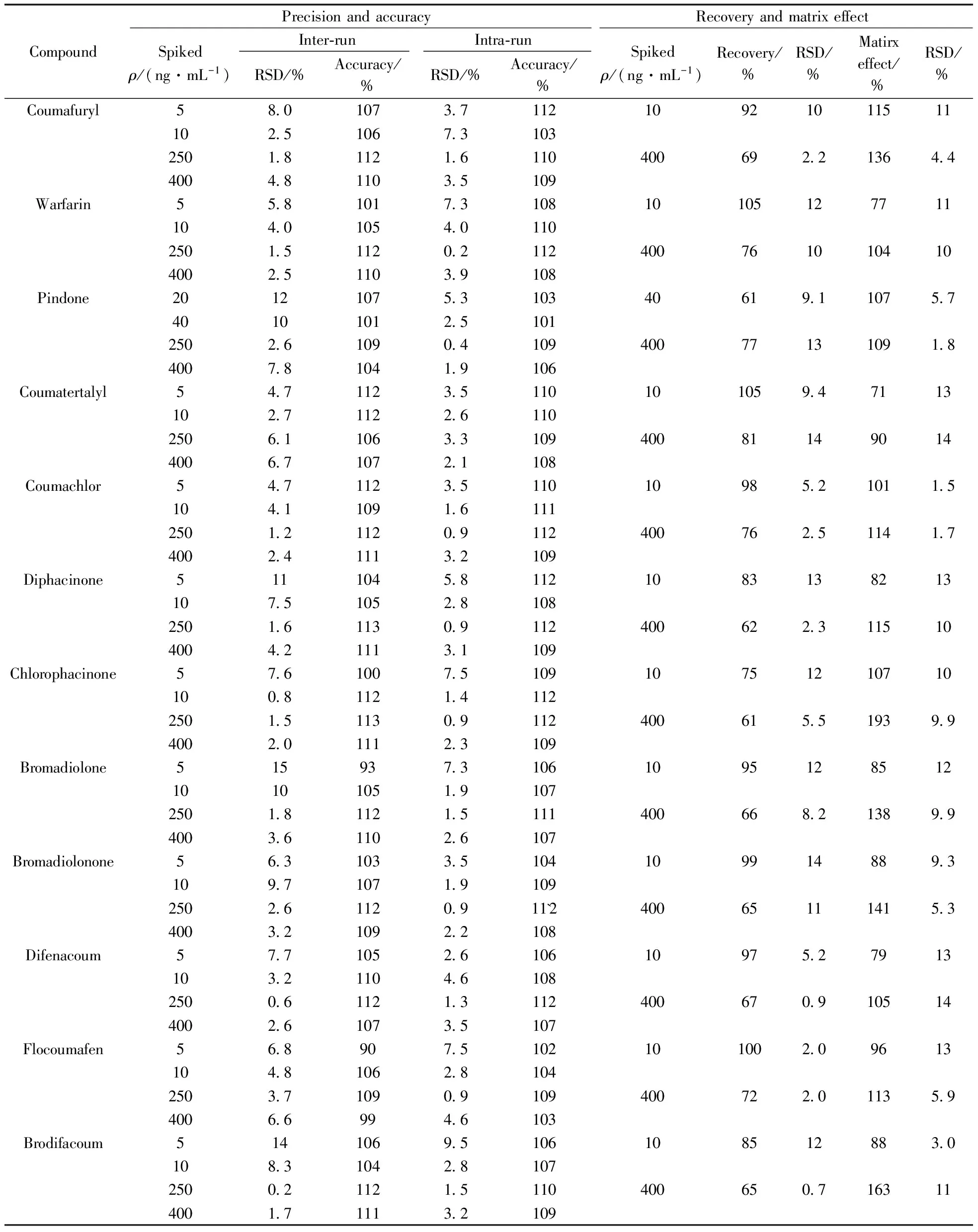
表3 干血斑中12种抗凝血杀鼠药的批内和批间准确度、精密度、回收率和基质效应Table 3 Inter- and intra-batch accuracy and precision,recovery and matrix effect of 12 anticoagulant rodenticides in dried blood spots
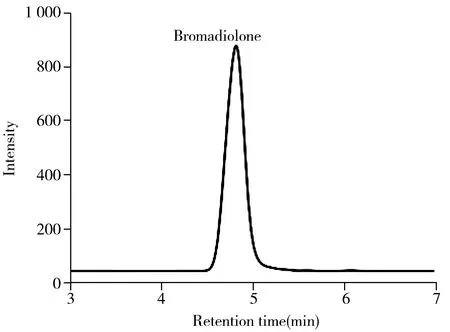
图4 患者干血斑样本中检测的抗凝血鼠药的MRM色谱图Fig.4 MRM chromatogram of anticoagulant rodenticides detected in the dried blood spotscoming from a clinical patient
2.5 应 用
本研究收集并制备了3位疑似抗凝血鼠药中毒患者的干血斑样本。患者在送医时,分别出现牙龈出血、尿血、鼻出血、尿色深等症状。经询问,各患者口述均未接触抗凝血鼠药或药物,初步推测为误食或者投毒所致。采用上述建立的方法,在1名未经维生素K治疗的患者血液中检出溴敌隆,质量浓度为(1.62±0.02) μg/mL,未检出其它鼠药,其MRM色谱图见图4。在另两位经服用维生素K治疗患者血液中检出溴敌隆,质量浓度为(0.13±0.01) μg/mL和(0.66±0.02)μg/mL,与其它报道的中毒浓度相当[9,20-21]。
3 结 论
本研究针对临床干血斑样本,建立了以水润湿、甲醇提取等普适的前处理方法,以霉酚酸作为内标,采用液相色谱-三重四极杆串联质谱实现了对12种香豆素类和茚二酮类抗凝血杀鼠药的定性定量分析。结果表明,在提取过程中加入适量的水润湿滤纸卡,有助于目标化合物从纤维素链上游离出来并获得较高的提取回收率。DMPK-A、DMPK-B、DMPK-C和903等4种滤纸卡对于抗凝血鼠药的干血斑分析存在差异,以903滤纸卡为基底时,各鼠药提取率均较高且结果稳定。该方法成功应用于临床实际中毒样本的检测,满足临床中毒快检的需求,为抗凝血鼠药中毒的诊断救治提供了可靠的新技术方法。
[1] Wanger M,Tonoli D,Varesio E,Hopfgartner G.MassSpectrom.Rev.,2014,35(3):361-438.
[2] Sharma A,Jaiswal S,Shukla M,Lal J.DrugTestingAnal.,2014,6(5):399-414.
[3] Shokry E,Villanelli F,Malvagia S,Rosati A,Forni G,Funghini S,Ombrone D,Bona M D,Guerrini R,La Marca G.J.Pharm.Biomed.Anal.,2015,109(1):164-170.
[4] Zikanova M,Krijt J,Skopova V,Krijt M,Baresova V,Kmoch S.Clin.Biochem.,2014,48(1/2):2-7.
[5] Heinig K,Bucheli F,Hartenbach R,Gajate-Perez A.Bioanalysis,2010,2(8):1423-1435.
[6] John H,Willoh S,Hörmann P,Siegert M,Vondran A,Thiermann H.Anal.Chem.,2016,88(17):8787-8894.
[7] Perez J W,Pantazides B G,Watson C M,Thomas J D,Blake T A,Johnson R C.Anal.Chem.,2015,87(11):5723-5729.
[8] King N,Tran M H.TransfusionMed.Rev.,2015,29(4):250-258.
[9] Yan H.FudanUniv.J.:Med.Sci.(严慧.复旦大学学报:医学版),2011,38(4):361-366.
[10] Imran M,Shafi H,Wattoo S A,Chaudhary M T,Usman H F.ForensicSci.Int.,2015,253:94-102.
[11] Palazoglu M G,Tor E R,Holstege D M,Galey F D.J.Agric.FoodChem.,1998,46(10):4260-4266.
[12] Guan F Y,Ishii A,Seno H,Watanabe K,Kumazawa T,Suzuki O.J.Pharm.Biomed.Anal.,1999,21(1):179-185.
[13] Maurer H H,Arlt J W.J.Chromatogr.B,1998,714(2):181-195.
[14] Jin M C,Ren Y P,Xu X M,Chen X H.ForensicSci.Int.,2007,171(1):52-56.
[15] Vandenbroucke V,Desmet N,De Backer P,Croubels S.J.Chromatogr.B,2008,869(1/2):101-110.
[16] Fourel I,Hugnet C,Goy-Thollot I,Berny P.J.Anal.Toxicol.,2010,34(2):95-103.
[18] Luckwell J,Denniff P,Capper S,Michael P,Spooner N,Mallender P,Johnson B,Clegg S,Green M,Ahmad S,Woodford L.Bioanalysis,2013,5(21):2613-2630.
[19] Henion J,Oliveira R V,Chace D H.Bioanalysis,2013,5(20):2547-2565.
[20] Vindenes V,Karinen R,Hasvold I,Bernard J P,Mørland J G,Christophersen A S.J.ForensicSci.,2008,53(4):993-996.
[21] Bidny S,Gago K,David M,Duong T,Albertyn D,Gunja N.J.Anal.Toxicol.,2015,39(3):219-224.
Simultaneous Determination of Twelve Anticoagulant Rodenticides in Clinical Dried Blood Spots by Liquid Chromatography-Tandem Mass Spectrometry
WANG Yi-jun1,GUO Lei1,XU Bin1,CHEN Jia1,LI Jia-wei2,QIU Ze-wu2,XIE Jian-wei1*
(1.State Key Laboratory of Toxicology and Medical Countermeasures,Institute of Pharmacology and Toxicology,Academy of Military Medical Sciences,Beijing 100850,China;2.Department of Poisoning and Treatment,The Affiliated Hospital of Academy of Military Medical Sciences,Beijing 100071,China)
To address the fast detection need towards poisoned patients in clinic,a sensitive,accurate liquid chromatographic-tandem mass spectrometric method was developed here for the qualitative and quantitative determinations of twelve anticoagulant rodenticides in dried blood spots.For the thoroughly punched dried blood spots,conditions influencing the extraction efficiency,including the kind of filter paper card,the water-soaking step and the kind of extraction solvent were investigated.A C18column was employed as separation phase while ammonium acetate in water (5 mmol/L)-ammonium acetate in methanol(5 mmol/L) was used as mobile phase by gradient elution.Then the sample was detected using negative electrospray ionization under multiple reaction monitoring mode.The results showed that,with the 903 type filter card as supporting matrix and a water-soaking step,followed by an extraction with methanol solvent containing internal standard for 5 min,the extraction efficiencies of the twelve rodenticides ranged from 66%to 115%with their intra-day relative standard deviations less than 15%.Good method validation results were obtained for all the anticoagulant rodenticides.The linearity for pindone was from 20 to 500 ng/mL with a correlation coefficient(r2) of 0.998 7,and that for the other eleven analytes were from 5 to 500 ng/mL with theirr2of 0.998 8-0.999 6.The recoveries for all the analytes ranged from 61%to 105%,with the matrix effects of 71%-193%and the intra-day relative standard deviations less than 15%.The established method was accurate,sensitive,rapid and convenient,and was successfully applied in the rapid detection of rodenticides in three clinic blood samples.It provides a new approach for clinical diagnosis and forensic toxicant analysis on anticoagulant rodenticide intoxication accidents.
dried blood spot;anticoagulant rodenticide;liquid chromatography-tandem mass spectrometry;fast detection on poisoned clinical samples
2017-06-20;
2017-07-20
全军医学科技“十三五”重大项目(AWS15J007))
*
谢剑炜,博士,研究员,研究方向:体内外毒物分析、质谱分析,Tel:010-66931649,E-mail:xiejw@bmi.ac.cn
10.3969/j.issn.1004-4957.2017.11.002
O657.7;O657.63
A
1004-4957(2017)11-1296-08
猜你喜欢
罕少疾病杂志(2022年9期)2022-09-03
医学理论与实践(2021年24期)2022-01-04
文萃报·周二版(2020年19期)2020-06-24
检验医学(2020年3期)2020-04-21
学苑创造·C版(2019年8期)2019-08-09
海外文摘·文学版(2018年2期)2018-02-21
东方剑(2017年9期)2017-11-13
刑事技术(2016年4期)2016-12-22
灾害医学与救援(电子版)(2016年3期)2016-03-11
中国卫生标准管理(2015年3期)2016-01-14
