
超声波合成1-对羟基苄基-2-对羟基苯基苯并咪唑
2017-11-23 06:20:16陈书君李敬芬
湖州职业技术学院学报 2017年3期
陈书君 , 李敬芬
(湖州师范学院 生命科学学院, 浙江 湖州 313000)
超声波合成1-对羟基苄基-2-对羟基苯基苯并咪唑
陈书君 , 李敬芬
(湖州师范学院 生命科学学院, 浙江 湖州 313000)
以氯化铵为催化剂,邻苯二胺和对羟基苯甲醛为原料,在超声波辐射下合成1,2-二取代的苯并咪唑,并利用正交实验筛选出最佳反应条件。通过对实验的探索得最佳反应条件为:配料比为2.3∶1.0,反应时间为2 h,超声波功率为140W,反应温度为55℃,氯化铵为催化剂40mmol,收率为79.06%。
超声波; 苯并咪唑; 邻苯二胺; 对羟基苯甲醛
苯并咪唑类化合物是一类苯并杂环化合物。多数苯并咪唑类化合物具有生物活性和活性反应, 可以作为药物中间体,在抗癌、抗真菌、止痛消炎等方面有重要的实用价值[1]432-435 [2]359-362。传统的1-对羟基苄基-2-对羟基苯基苯并咪唑的合成方法往往需要较为苛刻的条件,对设备的腐蚀性也较大,不符合绿色环保的理念[3]97-99 [4]909-912 [5]603-609 [6]423-427 [7]792-796。有学者通过微波合成该类化合物,但相对微波合成而言,超声波合成可以在接近室温或稍高于室温的较低温度下完成,安全性更佳[1]432-435 [5]603-609 [6]423-427。为此,本文试图探索一种简便高效的合成方法:以氯化铵作为催化剂,超声波为辅助催化手段,以无水乙醇作为溶剂,采用对羟基苯甲醛和邻苯二胺为原料,合成1-对羟基苄基-2-对羟基苯基苯并咪唑,以提高实验的安全性,降低原材料的消耗,提高收率,做到绿色排放。
一、实验部分
(一)仪器与试剂
本实验所用的仪器设备主要有:KQ5200DE型超声波清洗仪,WRS—1A型数字熔点仪,SHZ—DIII型循环式真空泵,BS124S型电子分析天平,1730型红外分光光度仪。所用的试剂主要有:对羟基苯甲醛(化学纯CP),邻苯二胺(化学纯CP),无水乙醇(分析纯AR),氯化铵(化学纯CP),其余试剂均为分析纯。
(二)合成方法
在100 mL圆底烧瓶中加入2.442 4 g对羟基苯甲醛和1.081 4 g邻苯二胺,而后加入35 mL无水乙醇,充分溶解后,再加入催化剂氯化铵,连接好回流装置并将圆底烧瓶放入超声波清洗仪中,设定好超声波反应的温度和功率及时间。待反应完毕后,冷却静置6 h,抽滤,洗涤,干燥,即得粗品。将粗品加入到50 mL烧杯中,加入30 mL无水乙醇,将烧杯置于热水浴中,至粗品完全溶解后加入少许活性炭脱色,用热滤漏斗趁热过滤除去氯化铵和杂质,滤液置于冰水浴中冷却至有结晶析出,抽滤,洗涤,干燥,得到最终产物。
二、实验结果与讨论
(一)正交优化实验
为了研究不同反应物摩尔比,反应温度,催化剂氯化铵的量对产率的影响,设计了以下的实验。
正交试验采用Ln(tq)表,L表示正交设计,t表示水平数,q表示因素数,n表示试验次数。选取超声波输出功率、原料摩尔比、反应温度、催化剂氯化铵的量作为四因素,进行四因素三水平正交设计实验。反应投料比n(对羟基苯甲醛)∶n(邻苯二胺)(A);因素安排为超声波功率(B),W;反应温度(C),℃;催化剂氯化铵的量(D)mmol。以产率为指标进行实验。表1为正交实验因素水平表(参见表1)。

表1 正交实验因素水平表
在本实验中选用的正交表格为L9(34),实验安排与收率如下表所示(参见表2)。
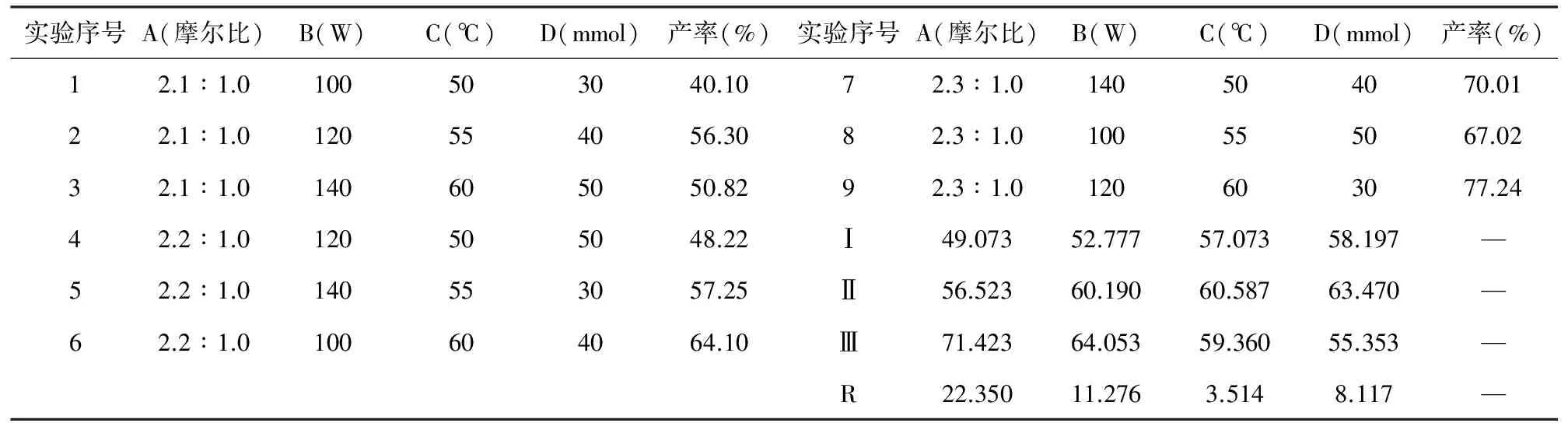
表2 L9(34)正交实验安排及实验结果
通过比较九个实验的转化率得出,第9号实验的转化率最高。进一步的分析,比较每一列的均值大小。影响大小顺序为A>B>D>C,第一列A的三号位级优于二号优于一号,第二列B的第三号位级优于二号优于一号位级,第三列C的二号位级优于三号优于一号,第四列D的第二位级优于一号优于二号位级。可以得出较好的条件是A3B3C2D2,当A因素配料比为2.3∶1.0,B因素超声波功率为140 W,C因素温度为55 ℃时,D因素为催化剂氯化铵40 mmol时,产物收率最高。
(二)优化条件下的重复实验结果
在正交设计所得的最佳条件下进行重复实验。实验结果见下表(参见表3)。
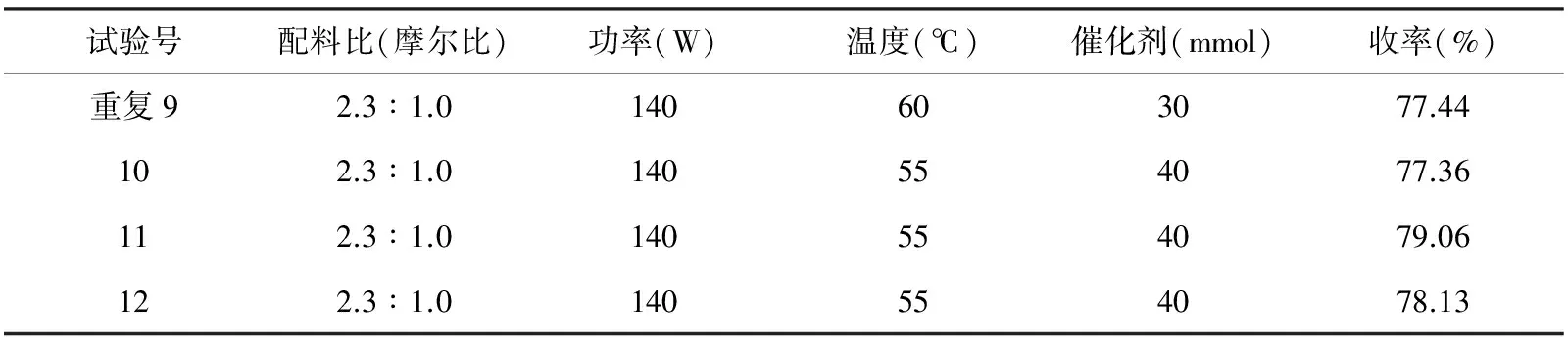
表3 正交设计最佳反应条件下重复实验结果表
由重复实验得出,最佳实验条件为重复实验的条件,测得1-对羟基苄基-2-对羟基苯基苯并咪唑的平均收率为78.18%,比实验9的产率高,且三次重复实验的结果相差很小,该方法的稳定性较好,收率客观。先用这种方法来合成目标产物。
(三)1-对羟基苄基-2-对羟基苯基苯并咪唑结构表征
采用压片法,将样品研细后和溴化钾粉末混研,待样品与溴化钾混合均匀,装入模具内用压片机压2~3分钟,压成透明的薄片,打开软件OPUS,设置参数,设置保存路径,扫描背景,将压好的片放入样品池,关上样品池,开始测试。图1为实验所得的红外光谱图(参见图1)。
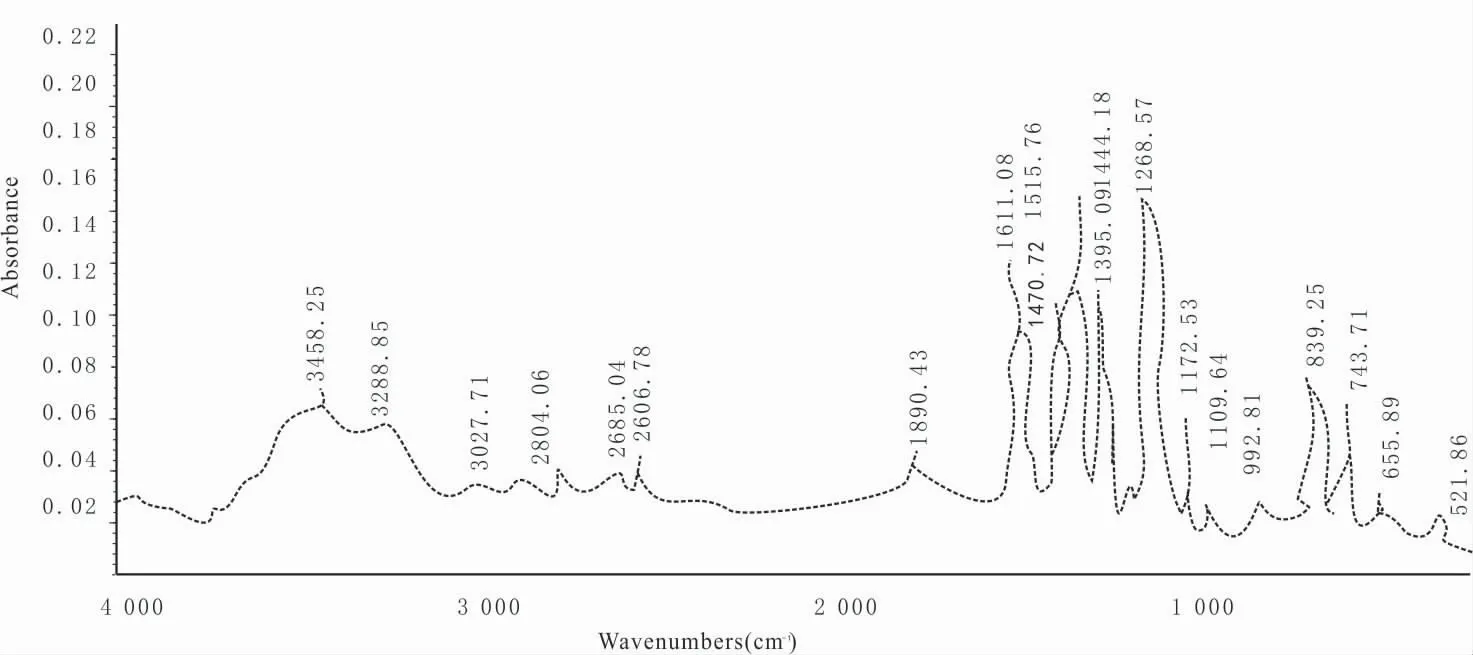
图1 1-对羟基苄基-2-对羟基苯基苯并咪唑红外光谱图
实验结果显示,产物的红外光谱主要特征峰为:3 288.85cm-1为γO-H(Ph-OH);3 027.71cm-1为γC-H(Ph);743.71cm-1为γC-H(O-Ph);839.25cm-1为γC-H(p-Ph);1 268.57cm-1为γC-N(Ph);1 515.76cm-1为γC=N(Imid),红外结果与标准谱图[7]792-796基本一致。结合所得产物颜色、结晶形状与文献基本符合,产物熔点为222.3~222.7℃,与文献报道的222.2~222.8℃基本一致[6]。基本确定该物质为1-对羟基苄基-2-对羟基苯基苯并咪唑。
三、结论与讨论
(一)结论
实验表明超声波可以加速二取代并咪唑合成的反应,并且不需要复杂的实验操作和苛刻的实验条件,超声波提供了一种绿色高效的合成方式。加入氯化铵可以提高反应的进行速度,提高产率。经过正交实验分析之后得出:在超声波辐射下,配料比为2.3∶1.0,反应时间为2 h,反应温度为60 ℃,超声波功率为140 W,氯化铵为催化剂40 mmol,收率为79.06%。
(二)讨论
在本实验中超声波反应时间过长会导致反应物被氧化,当反应时间超过6 h的时候产物被氧化变色,故最佳反应时长为2 h,不能反应过久。反应过程中会有少量的水生成,氯化铵会溶解一部分在水中,随着产物生成,反应物浓度降低反应速度下降。
在反应结束后需要静置6 h使产物充分析出,静置时间若少于6 h会使产物无法充分析出,静置时间过长则会被空气氧化。重结晶时不能用太多乙醇,30~35 ml为宜,乙醇过多会使析出变得缓慢。
[1] 吕维忠,刘 波,韦少慧.微波合成1-对羟基苄基-2-对羟基苯基苯并咪唑[J].深圳大学学报理工版,2007(4).
[2] 白翠冰,李军舰,张 鹏,等.合成苯并咪唑类化合物的工艺改进[J].合成化学,2012(3).
[3] 李莹莹,周永花,郭玉芳,等.苯并咪唑衍生物的合成改进[J].有机化学,2006(8).
[4] CARPENTER R D,DEBERDT P B,LAM K S.Synthesis and Potent Aimimicrobial Activities of Some Nqovel R:EtinoidalMonocation
icBenzimidazoles[J].Comb.Chem.,2006(5).
[5] 张守民,李 鸿,郑修成,等.超声波有机反应中的应用[J].有机化学,2002(9).
[6] 王 军,李军舰,初红涛,等.微波辐射下2-苯并咪唑的绿色合成[J].化学研究与应用,2013(3).
[7] 杨红伟,岳 凡,封 顺,等.苯并咪唑化合物一步法合成及表征[J].有机化学,2004(7).
Synthsisof1-hydroxy-benzyl-2-hydroxylPhenylBenzimidazolebyUnderUltrasonicIrradiation
CHEN Shu-jun , LI Jing-fen
(School Life Science, Huzhou University, Huzhou 313000, China )
The 1,2-disubstitutedbenzimidazole was synthesized by ultrasonic irradiation with ammonium chloride as catalyst, o-phenylenediamine and p - hydroxybenzaldehyde as reactants, and the optimum reaction conditions were screened by orthogonal experiment.The optimum reaction conditions were as follows: the ratio of ingredients was 2.3: 1.0, the reaction time was 2 h, the ultrasonic power was 140 W, the reaction temperature was 55 ℃, the ammonium chloride was 40 mmol, and the yield was 79.06%.
ultrasonic irradiation; benzimidazole; o-phenylenediamine; p-hydroxybenzaldehyde
2017-06-15
本文系2016年度湖州师范学院大学生创新创业训练计划重点项目“超声波合成1-对羟基苄基-2-对羟基苯基苯并咪唑”(153)的研究成果。
陈书君(1996-),女,浙江德清人,湖州师范学院生命科学学院2014级制药工程专业学生,主要从事有机物合成研究;李敬芬(1964-),女,黑龙江绥化人,教授,化学硕士,主要从事有机合成、天然有效成分结构修饰技术研究。
O644.3
A
1672-2388(2017)03-0089-03
猜你喜欢
中国资源综合利用(2017年2期)2018-01-22 02:44:58
化工环保(2017年5期)2017-10-31 07:04:21
新乡学院学报(2016年6期)2016-12-01 05:21:38
合成化学(2015年2期)2016-01-17 09:03:42
合成化学(2015年10期)2016-01-17 08:55:42
合成化学(2015年1期)2016-01-17 08:53:55
分析科学学报(2015年3期)2015-10-18 02:25:54
中国洗涤用品工业(2015年9期)2015-02-28 19:03:05
郑州大学学报(理学版)(2014年4期)2014-03-01 04:21:21
无机化学学报(2014年8期)2014-02-28 17:32:46
