
发作性运动诱发性运动障碍临床表型分析
2017-11-21 11:46:23田沃土黄啸君沈隽逸徐洋奇陈生弟曹立
中国现代神经疾病杂志 2017年7期
田沃土 黄啸君 沈隽逸 徐洋奇 陈生弟 曹立
.神经系统遗传性疾病.
发作性运动诱发性运动障碍临床表型分析
田沃土 黄啸君 沈隽逸 徐洋奇 陈生弟 曹立
研究背景发作性运动诱发性运动障碍是一组由突然动作诱发的非随意性运动障碍性疾病,表现为反复发作的短暂性肌张力障碍或舞蹈样动作,具有高度临床和遗传异质性.本研究旨在总结中国发作性运动诱发性运动障碍临床表型特点.方法采集195例原发性发作性运动诱发性运动障碍患者临床资料,采用自行设计的发作性运动诱发性运动障碍登记表记录并整理,分析和总结发作性运动诱发性运动障碍临床表型特点,并比较家族性与散发性患者临床表型差异.结果195例发作性运动诱发性运动障碍患者男女比例为4.42∶1,平均发病年龄为(12.32±3.49)岁,单纯型162例(83.08%)、复杂型33例(16.92%),16例(8.21%)合并特发性震颤,144例(73.85%)发作前有先兆,发作形式包括肌张力障碍(134例,68.72%)、舞蹈样动作(8例,4.10%)和二者混合形式(53例,27.18%),134例(68.72%)发作时面部受累,115例(58.97%)发作频率<10次/d、54例(27.69%)10~20次/d、26例(13.33%)>20~30 次/d,117例(60%)发作持续时间<10 s、58例(29.74%)>10~30 s、20例(10.26%)>30~60 s,散发性131例(67.18%)、家族性64例(32.82%),78例(40%)未服用药物,117例(60%)服用抗癫药物患者中106例症状完全控制、8例偶有发作、3例未见明显缓解.其中,家族性组发病年龄低于(t=2.376,P=0.019)、发作持续时间短于(χ2=7.731,P=0.021)散发性组.结论通过大样本临床数据分析和总结中国发作性运动诱发性运动障碍临床表型特点,以期为临床诊断与治疗提供帮助.
运动障碍; 表型
发作性运动诱发性运动障碍[PKD,在线人类孟德尔遗传数据库(OMIM)编号:128200]是发作性运动障碍的最常见类型,系一组突然动作诱发的非随意性运动障碍性疾病,发作时以异常运动或姿势为特征,如肌张力障碍、舞蹈样动作、手足徐动、投掷样动作等,可持续数秒至数十秒,发作间期正常[1].自2011年首个致病基因PRRT2基因克隆以来,发作性运动诱发性运动障碍在分子生物学和功能影像学等领域取得显著进展[2⁃8],逐渐受到临床医师和研究者们的关注.本研究回顾分析195例发作性运动诱发性运动障碍患者的临床资料,总结中国发作性运动诱发性运动障碍的临床表型特点.
对象与方法
一、研究对象
选择2008年8月-2016年12月在上海交通大学医学院附属瑞金医院神经科就诊的原发性发作性运动诱发性运动障碍患者共195例,均符合原发性发作性运动诱发性运动障碍诊断标准[1]:由突然动作诱发;发作持续时间短暂(<1 min);发作期意识清晰;发病年龄1~20岁,如有家族史,发病年龄适当增宽;苯妥英钠或卡马西平可以有效控制癫发作;神经系统检查和神经电生理学检查正常;同时能够配合完整的神经系统查体和详细的病史采集.排除其他继发性因素,如癫、多发性硬化(MS)、中枢神经系统感染、代谢性疾病(如甲状腺功能减退症或甲状腺功能亢进症)、心源性运动障碍等.本研究经上海交通大学医学院附属瑞金医院道德伦理委员会审核批准,所有患者及其家属均知情同意并签署知情同意书.
二、研究方法
1.临床资料采集 采用上海交通大学医学院附属瑞金医院神经科自行设计的发作性运动诱发性运动障碍登记表,由神经科医师和研究生记录并整理发作性运动诱发性运动障碍患者的临床资料,包括性别,年龄,发病年龄,发作类型,婴儿惊厥、偏头痛、发作性共济失调等其他发作性疾病病史,合并症,先兆比例,发作形式,累及部位,面部受累,发作频率,发作持续时间,家族史,自愈倾向等,将上述临床资料录入计算机并建立数据库,比较家族性与散发性发作性运动诱发性运动障碍临床表型特点.
2.统计分析方法 采用SPSS 16.0统计软件进行数据处理与分析.计数资料以相对数构成比(%)或率(%)表示,采用χ2检验;计量资料以均数±标准差(x±s)表示,采用两独立样本的t检验.以P≤0.05为差异具有统计学意义.
结 果
一、发作性运动诱发性运动障碍的临床表型
195例发作性运动诱发性运动障碍患者,男性159例,女性36例;年龄8~65岁,平均(29.43±13.41)岁;发病年龄4个月至27岁,平均(12.32±3.49)岁,其中161例(82.56%)于7~15岁(青少年期)发病;单纯型发作性运动诱发性运动障碍162例(83.08%),复杂型发作性运动诱发性运动障碍33例(16.92%),其中19例有婴儿惊厥病史、13例有偏头痛病史、1例有发作性共济失调病史;合并特发性震颤16例(8.21%);所有患者(195例,100%)均由突然动作诱发,亦可由突然加速(137例,70.26%)和意图动作(67例,34.36%)诱发,其中114例(58.46%)和36例(18.46%)分别于情绪紧张和疲劳状态下症状加重;144例(73.85%)发作前有先兆,主要来自单侧或双侧肢体远端并向近端蔓延;发作形式包括肌张力障碍(134例,68.72%)、舞蹈样动作(8例,4.10%)和二者混合形式(53例,27.18%);发作时80例(41.03%)累及单侧肢体,43例(22.05%)累及双侧肢体,49例(25.13%)累及单侧或双侧肢体,23例(11.79%)双侧交替发作;134例(68.72%)发作时有面部障碍,表现为表情僵硬、瞪眼、龇牙、咧嘴和构音障碍等;115例(58.97%)发作频率<10次/d,54例(27.69%)发作频率为10~20次/d,26例(13.33%)发作频率>20~30次/d;117例(60%)发作持续时间<10 s、58例(29.74%)10~30 s、20例(10.26%)>30~60 s;散发性发作性运动诱发性运动障碍131例(67.18%),家族性发作性运动诱发性运动障碍64例(32.82%);83例(42.56%)完全或不完全自愈,自愈年龄19~34岁、平均(21.51±4.42)岁;78例(40%)未服用药物,117例(60%)服用抗癫药物(AEDs)患者中89例予卡马西平、17例予奥卡西平、11例予其他抗癫药物(如苯妥英钠、托吡酯、丙戊酸钠、拉莫三嗪和氯硝西泮);服用卡马西平(50~100 mg/d)的89例患者中86例完全控制、2例部分控制、1例未见明显缓解,服用奥卡西平(75~150 mg/d)的17例患者中13例症状完全控制、4例部分控制,总体而言,服用抗癫药物的117例患者中106例(90.60%)症状完全控制、8例(6.84%)发作频率下降但偶有发作、3例(2.56%)未见明显缓解(1例予卡马西平,2例予丙戊酸钠).
二、家族性与散发性发作性运动诱发性运动障碍的临床表型比较
195例发作性运动诱发性运动障碍患者根据是否有家族史分为家族性发作性运动诱发性运动障碍组(家族性组)和散发性发作性运动诱发性运动障碍组(散发性组).(1)家族性组:64例患者,男性48例,女性16例;年龄8~65岁,平均(30.09±11.55)岁;发病年龄4个月至27岁,平均(11.48±3.83)岁;单纯型发作性运动诱发性运动障碍54例(84.38%),复杂型发作性运动诱发性运动障碍10例(15.63%),其中3例有婴儿惊厥病史、7例偏头痛病史;合并特发性震颤6例(9.38%);44例(68.75%)发作前有先兆;发作形式表现为肌张力障碍50例(78.13%)、舞蹈样动作2例(3.13%)和二者混合形式12例(18.75%);发作时25例(39.06%)累及单侧肢体,12例(18.75%)累及双侧肢体,14例(21.88%)累及单侧或双侧肢体,13例(20.31%)双侧交替发作;47例(73.44%)发作时累及面部;39例(60.94%)发作频率 <10次/d,17例(26.56%)10~20次/d,8例(12.50%)>20~30次/d;47例(73.44%)发作持续时间 <10 s、14例(21.88%)为10~30 s、3例(4.69%)>30~60 s;39例(60.94%)未服用药物,25例(39.06%)服用抗癫药物患者中23例症状完全控制、2例部分控制.(2)散发性组:131例患者,男性111例,女性20例;年龄9~41岁,平均(23.70±18.01)岁;发病年龄6~23岁,平均(12.73±3.25)岁;单纯型发作性运动诱发性运动障碍108例(82.44%),复杂型发作性运动诱发性运动障碍23例(17.56%),其中16例有婴儿惊厥病史、6例偏头痛病史、1例发作性共济失调病史;合并特发性震颤10例(7.63%);100例(76.34%)发作前有先兆;发作形式表现为肌张力障碍84例(64.12%)、舞蹈样动作6例(4.58%)和二者混合形式41例(31.30%);发作时55例(41.98%)累及单侧肢体,31例(23.66%)累及双侧肢体,35例(26.72%)累及单侧或双侧肢体,10例(7.63%)双侧交替发作;87例(66.41%)发作时累及面部;76例(58.02%)发作频率<10次/d,37例(28.24%)发作频率10~20次/d,18例(13.74%)>20~30次/d;70例(53.44%)发作持续时间 <10 s、44例(33.59%)10~30 s、17例(12.98%)>30~60 s;39例(29.77%)未服用药物,92例(70.23%)服用抗癫药物患者中83例症状完全控制、6例部分控制、3例未见明显缓解.由表1可见,两组患者临床表型比较,性别、发作类型、合并症、先兆比例、发作形式、累及部位、面部受累、发作频率、服用抗癫药物比例和药物疗效差异无统计学意义(均P>0.05),而家族性组发病年龄低于(P=0.019)、发作持续时间短于(P=0.021)散发性组且差异有统计学意义.
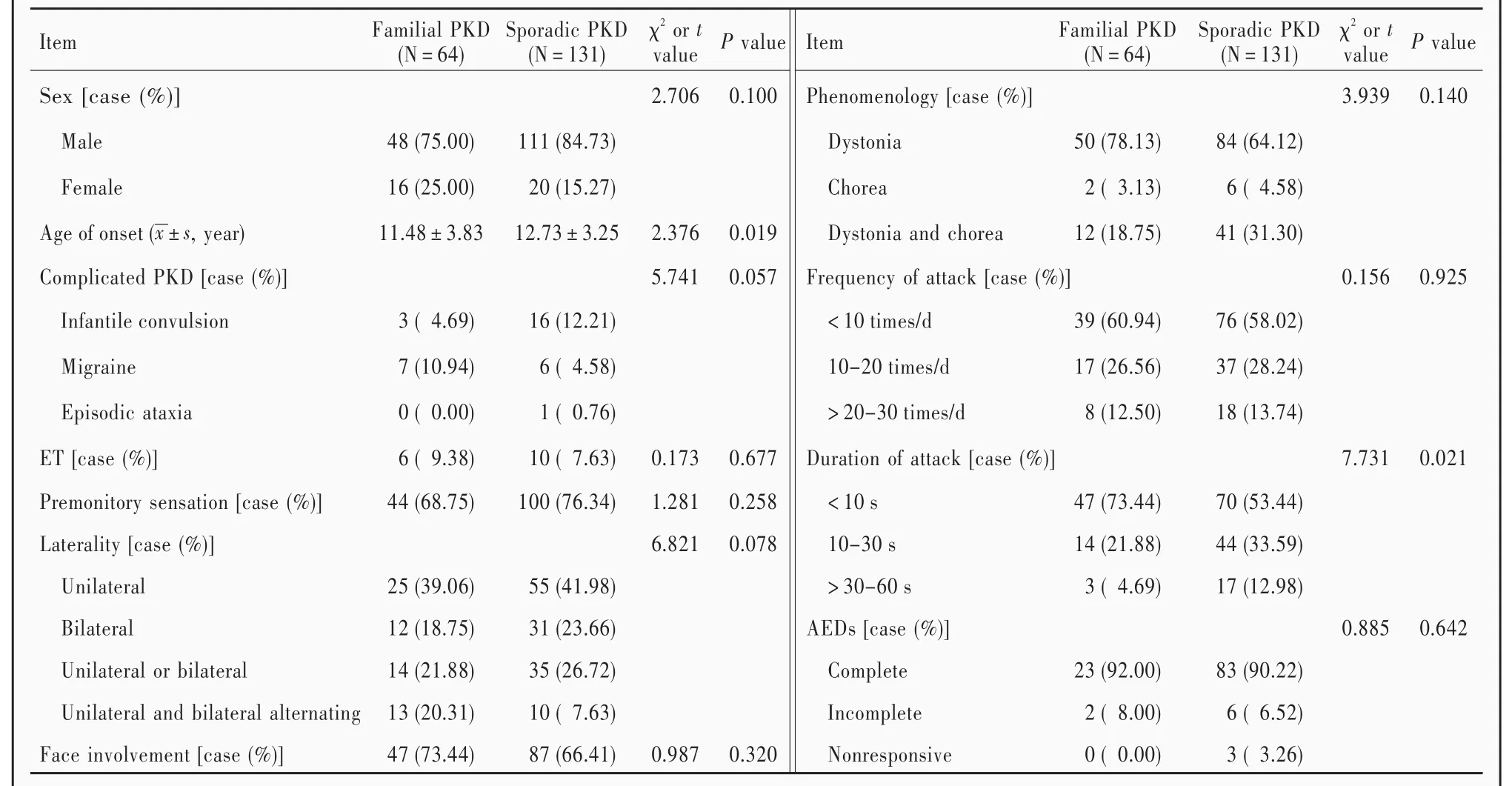
表1 家族性组与散发性组患者临床表型的比较Table 1. Comparison of general data between familial and sporadic PKD patients
讨 论
发作性运动障碍是一类以反复发作的非随意性运动障碍为主要特征的罕见神经系统疾病,具有高度临床异质性和遗传异质性.发作性运动诱发性运动障碍是发作性运动障碍的最常见类型[9],按照病因可以分为原发性和继发性,其中,原发性发作性运动诱发性运动障碍根据有无家族史又分为家族性和散发性,家族性发作性运动诱发性运动障碍呈常染色体显性遗传.2011年首次证实PRRT2基因是发作性运动诱发性运动障碍的致病基因之一,定位于16p11.2,包含4个外显子,热点突变为c.649dupC(p.Arg217ProfsX8)[2⁃3,10⁃11],编码富含脯氨酸跨膜蛋白2(PRRT2).PRRT2蛋白是由340个氨基酸组成的一次跨膜蛋白,其氨基末端(N末端)较长朝向胞内,羧基末端(C末端)较短朝向胞外[12].PRRT2蛋白表达于神经元突触,通过与突触结合蛋白1/2(Syt1/2)相互作用参与钙离子的快速识别,从而参与神经递质的释放过程[13⁃14].婴儿惊厥、良性家族性婴儿惊厥(BFIS)、婴儿惊厥伴阵发性舞蹈手足徐动症、家族性偏瘫型偏头痛、发作性共济失调、热性惊厥、偏头痛、发作性非运动诱发性运动障碍及家族性和散发性发作性过度运动诱发性运动障碍患者也存在PRRT2基因突变.提示上述发作性疾病包括发作性动作诱发性运动障碍可能属PRRT2基因相关疾病谱范畴,统称为PRRT2相关疾病(PRD)[15⁃18].在目前报道的病例中,约61.5%家族性发作性运动诱发性运动障碍为PRRT2基因突变所致,此外,尚有12.5%~89.5%散发性发作性运动诱发性运动障碍无PRRT2基因突变[5,19⁃20],提示存在除PRRT2基因外的其他可能致病基因.
在本研究中,发作性运动诱发性运动障碍患者发病年龄4个月至27岁,以7~15岁青少年为高发,男女比例4.42∶1,散发性病例男女比例5.55∶1.发作性运动诱发性运动障碍多由突然动作诱发,如起立、接电话或起跑等,运动速度和幅度改变以及意图动作或在持续动作中加入其他动作时可诱发,此外情绪紧张、疲劳等亦可诱发.73.85%患者发作前有发作预感,表现为受累肢体无力感、受累部位肌肉紧张感、浅感觉不一和头晕等.部分患者在先兆症状后通过减慢患肢动作以阻止发作.发作形式包括肌张力障碍、舞蹈样动作或混合发作,多为单侧发作,亦可双侧同时或交替发作.同一家系发作性运动诱发性运动障碍患者临床表现相似[15].有2/3患者发作时累及面部肌肉,出现挤眉弄眼和构音障碍.发作频率多于青春期达高峰,自发缓解年龄为20~30岁,部分患者30岁后很少发作甚至完全自愈.比较家族性与散发性发作性运动诱发性运动障碍患者的临床表型特点,发现家族性患者发病年龄较早、发作持续时间较短,而其他临床表现无差异.原发性发作性运动诱发性运动障碍应与癫、心源性运动障碍、癔症、特发性肌张力障碍、先天性肌强直等相鉴别,并排除可能的继发性因素,如颅脑创伤、中枢神经系统肿瘤、中枢神经系统感染、甲状腺疾病和自身免疫性疾病等[1,21⁃23].
综上所述,本研究从大样本水平描述和总结中国发作性运动诱发性运动障碍的临床表型特点,并比较家族性与散发性患者临床表型差异.在临床实践中,应进一步提高临床医师对发作性运动诱发性运动障碍的认识,重视临床医师(包括神经内科和儿科医师)的培训,明确诊断标准和鉴别诊断要点,帮助患者第一时间诊断,并使其明确经合理的药物治疗可以有效控制疾病,且随年龄的增长可能出现自发性缓解.尽管发作性运动诱发性运动障碍的发病机制以及临床表型与基因型的关系尚未明确,但随着研究的不断深入,其临床异质性和遗传异质性之谜将被解开,同时也将给新致病基因的发现和发病机制的研究提供新的视野.
[1]Bruno MK,Hallett M,Gwinn⁃Hardy K,Sorensen B,Considine E,Tucker S,Lynch DR,Mathews KD,Swoboda KJ,Harris J,Soong BW,Ashizawa T,Jankovic J,Renner D,Fu YH,Ptacek LJ.Clinical evaluation of idiopathic paroxysmal kinesigenic dyskinesia:new diagnostic criteria.Neurology,2004,63:2280⁃2287.
[2]Chen WJ,Lin Y,Xiong ZQ,Wei W,Ni W,Tan GH,Guo SL,He J,Chen YF,Zhang QJ,Li HF,Lin Y,Murong SX,Xu J,Wang N,Wu ZY.Exomesequencingidentifiestruncating mutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia.Nat Genet,2011,43:1252⁃1255.
[3]Wang JL,Cao L,Li XH,Hu ZM,Li JD,Zhang JG,Liang Y,San A,Li N,Chen SQ,Guo JF,Jiang H,Shen L,Zheng L,Mao X,Yan WQ,Zhou Y,Shi YT,Ai SX,Dai MZ,Zhang P,Xia K,Chen SD,Tang BS.Identification of PRRT2 as the causative gene of paroxysmal kinesigenic dyskinesias.Brain,2011,134:3493⁃3501.
[4]Ebrahimi⁃Fakhari D,Saffari A,Westenberger A,Klein C.The evolving spectrum of PRRT2⁃associated paroxysmal diseases.Brain,2015,138:3476⁃3495.
[5]Huang XJ,Wang T,Wang JL,Liu XL,Che XQ,Li J,Mao X,Zhang M,Bi GH,Wu L,Zhang Y,Wang JY,Shen JY,Tang BS,Cao L,Chen SD.Paroxysmalkinesigenic dyskinesia:clinical and genetic analyses of 110 patients.Neurology,2015,85:1546⁃1553.
[6]Valtorta F,BenfenatiF,Zara F,MeldolesiJ.Part2:from paroxysmal disorders to regulation of synaptic function.Trends Neurosci,2016,39:668⁃679.
[7]Hsu WY,Kwan SY,Liao KK,Chen RS,Lin YY.Altered inhibitory modulation of somatosensory cortices in paroxysmal kinesigenic dyskinesia.Mov Disord,2013,28:1728⁃1731.
[8]Hsiao FJ,Hsu WY,Chen WT,Chen RS,Lin YY.Abnormal somatosensory synchronization in patients with paroxysmal kinesigenic dyskinesia:a magnetoencephalographic study.Clin EEG Neurosci,2017,48:288⁃294.
[9]Erro R,Sheerin UM,Bhatia KP.Paroxysmaldyskinesias revisited:a review of 500 genetically proven cases and a new classification.Mov Disord,2014,29:1108⁃1116.
[10]van Vliet R,Breedveld G,de Rijk⁃van Andel J,Brilstra E,Verbeek N,Verschuuren⁃Bemelmans C,Boon M,Samijn J,Diderich K,van de Laar I,Oostra B,Bonifati V,Maat⁃Kievit A.PRRT2 phenotypes and penetrance of paroxysmal kinesigenic dyskinesia and infantile convulsions.Neurology,2012,79:777⁃784.
[11]Wu L,Tang HD,Huang XJ,Zheng L,Liu XL,Wang T,Wang JY,Cao L,Chen SD.PRRT2 truncated mutations lead to nonsense⁃mediated mRNA decay in paroxysmal kinesigenic dyskinesia.Parkinsonism Relat Disord,2014,20:1399⁃1404.
[12]Rossi P,Sterlini B,Castroflorio E,Marte A,Onofri F,Valtorta F,Maragliano L,Corradi A,Benfenati F.A novel topology of proline⁃rich transmembrane protein 2(PRRT2):hints foe an intracellular function at the synapse.J Biol Chem,2016,291:6111⁃6123.
[13]Valente P,Castroflorio E,RossiP,Fadda M,SterliniB,CervigniRI,Prestigio C,GiovediS,OnofriF,Mura E,Guarnieri FC,Marte A,Orlando M,Zara F,Fassio A,Valtorta F,BaldelliP,CorradiA,BenfenatiF.PRRT2 isa key component of the Ca(2+)⁃dependent neurotransmitter release machinery.Cell Rep,2016,15:117⁃131.
[14]Li M,Niu F,Zhu X,Wu X,Shen N,Peng X,Liu Y.PRRT2 mutant leads to dysfunction of glutamate signaling.Int J Mol Sci,2015,16:9134⁃9151.
[15]Becker F,Schubert J,Striano P,Anttonen AK,Liukkonen E,Gaily E,Gerloff C,Müller S,Heußinger N,Kellinghaus C,Robbiano A,Polvi A,Zittel S,Oertzen TJ,Rostasy K,Schöls L,Warner T,Munchau A,Lehesjoki AE,Zara F,Lerche H,Weber YG.PRRT2⁃related disorders:further PKD and ICCA cases and review of the literature.J Neurol,2013,260:1234⁃1244.
[16]Méneret A,Roze E.Paroxysmal movement disorders:an update.Rev Neurol(Paris),2016,172:433⁃445.
[17]Méneret A,Grabli D,Depienne C,Gaudebout C,Picard F,Dürr A,Lagroua I,Bouteiller D,Mignot C,Doummar D,Anheim M,Tranchant C,Burbaud P,Jedynak CP,Gras D,Steschenko D,Devos D,Billette de VillemeurT,Vidailhet M,Brice A,Roze E.PRRT2 mutations:a major cause of paroxysmal kinesigenic dyskinesia in the European population.Neurology,2012,79:170⁃174.
[18]Torisu H,Watanabe K,Shimojima K,Sugawara M,Sanefuji M,Ishizaki Y,Sakai Y,Yamashita H,Yamamoto T,Hara T.Girl with a PRRT2 mutation and infantile focalepilepsy with bilateral spikes.Brain Dev,2014,36:342⁃345.
[19]Heron SE,Dibbens LM.Role of PRRT2 in common paroxysmal neurological disorders:a gene with remarkable pleiotropy.J Med Genet,2013,50:133⁃139.
[20]Lee YC,Lee MJ,Yu HY,Chen C,Hsu CH,Lin KP,Liao KK,Chang MH,Liao YC,Soong BW.PRRT2 mutationsin paroxysmal kinesigenic dyskinesia with infantile convulsions in a Taiwanese cohort.PLoS One,2012,7:E38543.
[21]Thomas KP, Muthugovindan D, Singer HS. Paroxysmal kinesigenic dyskinesias and pseudohypo⁃parathyroidism type Ⅰb.Pediatr Neurol,2010,43:61⁃64.
[22]HuangXJ,CaoL,Chen SD.Progress in theresearch of genetics and clinical manifestation of paroxysmal kinesigenic dyskinesia.Zhongguo Xian Dai Shen Jing Ji Bing Za Zhi,2013,13:457⁃462[.黄啸君,曹立,陈生弟.发作性动作诱发性运动障碍临床表现及遗传学研究进展.中国现代神经疾病杂志,2013,13:457⁃462.]
[23]Wang JD,Bi GR,Cong SY,Zheng DM,Xu FF.Four cases of paroxysmal kinesigenic dyskinesia.Zhongguo Xian Dai Shen Jing Ji Bing Za Zhi,2009,9:309⁃310[.王建东,毕国荣,丛树艳,郑东明,徐菲菲.发作性运动诱发舞蹈手足徐动症四例.中国现代神经疾病杂志,2009,9:309⁃310.]
Clinical phenotype analysis of paroxysmal kinesigenic dyskinesia
TIAN Wo⁃tu1,HUANG Xiao⁃jun2,SHEN Jun⁃yi1,XU Yang⁃qi1,CHEN Sheng⁃di1,CAO Li11Department of Neurology and Institute of Neurology,Ruijin Hospital,School of Medicine,Shanghai Jiaotong University,Shanghai 200025,China
2Department of Neurology,North Department of Ruijin Hospital,School of Medicine,Shanghai Jiaotong University,Shanghai 201801,China
CAO Li(Email:caoli2000@yeah.net)
BackgroundParoxysmal kinesigenic dyskinesia(PKD)is a disorder characterized by recurrent and brief dystonic or choreoathetoid attacks that are induced by sudden voluntary movement with highly clinical and genetic heterogeneity.We aimed to investigate the clinical features of PKD in a large Chinese population.MethodsOne hundred and ninety five patients diagnosed as primary PKD were recruited.For all of the participants,neurological examinations were conducted and clinical manifestations were recorded and summarized in self⁃made uniform registration form for PKD patients. Clinical characteristics were statistically analyzed and compared between familial and sporadic PKD patients.ResultsAmong all of the 195 PKD patients in the present study,the gender ratio was 4.42∶1(male∶female).The average age of onset was(12.32±3.49)years.There were 162 patients(83.08%)manifestated with pure form and 33(16.92%)with complicated form of PKD.Among them 16 patients(8.21%)had essential tremor(ET),and 144 patients(73.85%)had premonitory symptom.The percentage of patients manifested as dystonia,chorea and mixed form during episodic attacks were 68.72%(134/195),4.10%(8/195)and 27.18%(53/195)repectively.There were 134 cases(68.72%)had facial involvement.It was recorded that 115(58.97%),54(27.69%)and 26(13.33%)patients had frequency of attack<10 times/d,10-20 times/d and>20-30 times/d respectively. The percentages of patients whose duration of attack<10 s,10-30 s and>30-60 s were 60%(117/195),29.74%(58/195)and 10.26%(20/195)respectively.There were 64 patietns(32.82%)with family history of PKD and 131(67.18%)were sporadic PKD patients.Up to 40%(78/195)of patients did not require/take medications,as they had minor clinical manifestations or concerns about the side effects of anticonvulsants. Among 117 patients(60%)prescribed with anticonvulsants,114 patients showed a good response,including complete control(N=106)and partial control(N=8),and 3 patients were nonresponsive.In comparison with sporadic PKD patients,familial PKD patients had earlier age of onset(t=2.376,P=0.019)and shorter duration of attack(χ2=7.731,P=0.021)respectively.ConclusionsWe summarized the clinical characteristics of PKD patients in mainland China.Through the analysis of large sample data,we hope to improve and standardize the diagnosis and treatment of PKD clinically.
Movement disorders; Phenotype
10.3969/j.issn.1672⁃6731.2017.07.006
国家自然科学基金资助项目(项目编号:81571086);国家自然科学基金资助项目(项目编号:81271262);国家自然科学基金青年科学基金资助项目(项目编号:81600978);上海交通大学医学院高峰高原计划(项目编号:20161401);上海交通大学"医工交叉研究基金"资助项目(项目编号:YG2016MS64);上海交通大学医学院"大学生创新训练计划"(项目编号:2015045)
200025上海交通大学医学院附属瑞金医院神经科 上海交通大学医学院神经病学研究所(田沃土,沈隽逸,徐洋奇,陈生弟,曹立);201801上海交通大学医学院附属瑞金医院北院神经内科(黄啸君)
曹立(Email:caoli2000@yeah.net)
This study was supported by the National Natural Science Foundation of China(No.81571086,81271262),the National Natural Science Foundation of China for Young Scientists(No.81600978),Shanghai Jiaotong University School of Medicine Peak and Plateau Program(No.20161401),Crossing Program between Medicine and Industry supported by Shanghai Jiaotong University(No.YG2016MS64),and Shanghai Jiaotong University School of Medicine Undergraduate Innovation Training Program(No.2015045).
2017⁃06⁃20)
猜你喜欢
中国神经精神疾病杂志(2022年3期)2022-07-14 02:23:42
世界科学技术-中医药现代化(2021年8期)2021-12-21 07:04:52
中国医药导报(2020年16期)2020-07-31 09:43:38
读天下(2020年2期)2020-04-14 04:48:48
现代园艺(2017年21期)2018-01-03 06:41:32
中国医药科学(2015年24期)2016-03-07 15:32:46
淮海医药(2015年1期)2016-01-12 04:32:54
中国康复理论与实践(2015年10期)2015-12-24 05:42:44
医学研究杂志(2015年5期)2015-06-10 06:43:26
现代检验医学杂志(2015年5期)2015-02-06 01:42:20
