
共济失调毛细血管扩张症四例临床表型及基因突变分析
2017-11-21 11:46:24郑岚刘晓黎曹立
中国现代神经疾病杂志 2017年7期
郑岚 刘晓黎 曹立
共济失调毛细血管扩张症四例临床表型及基因突变分析
郑岚 刘晓黎 曹立
目的报道4例经基因检测明确诊断的共济失调毛细血管扩张症患者,结合文献总结该病临床表型和基因突变特点.方法采集3个共济失调毛细血管扩张症家系共4例患者临床资料,并提取患者及其父母外周静脉血,采用全外显子测序和Sanger测序进行ATM基因突变分析.结果4例共济失调毛细血管扩张症患者均表现为儿童期发病的进行性进展的小脑共济失调、球结膜和皮肤毛细血管扩张、免疫缺陷导致反复感染,血清甲胎蛋白水平升高,头部MRI显示小脑萎缩.ATM基因检测显示,例1和例2存在已知复合杂合突变c.8287C>T(p.Arg2763X)和c.9139C>T(p.Arg3047X),均为无义突变;例3存在2种未报道的复合杂合突变,包括无义突变c.8911C>T(p.Gln2971X)和缺失突变c.7141_7151delAATGGAAAAAT(p.Asn2381GlufsX18);例 4存在纯合突变 c.1402_1403delAA(p.Lys468GlufsX18).结论4例患者具有典型共济失调毛细血管扩张症临床表现.变异型共济失调毛细血管扩张症患者神经系统受累较轻,头部MRI通常正常,神经系统以外受累少见,明确诊断仍依靠ATM基因检测.
共济失调性毛细血管扩张; 表型; 基因; 突变
共济失调毛细血管扩张症[AT,在线人类孟德尔遗传数据库(OMIM)编号:208900]亦称Louis⁃Bar综合征,是常染色体隐性遗传性多系统疾病,系ATM基因突变(OMIM编号:607585)所致,累及神经系统、免疫系统、内分泌系统和皮肤等[1⁃2].该病全球发病率1/10万~1/4万,通常于婴幼儿期(1~5岁)发病,临床主要表现为进行性小脑共济失调,球结膜和面部毛细血管扩张,反复发作的呼吸道感染[3],其中,神经系统受累还可以表现为吟诗样语言、肌阵挛、肌张力障碍、眼运动性共济失调等;实验室检查血清甲胎蛋白(AFP)水平升高和染色体不稳定;头部MRI显示小脑萎缩;疾病晚期易合并恶性肿瘤.本研究报道4例经基因检测明确诊断的共济失调毛细血管扩张症患者,并复习相关文献,总结该病临床表型和基因突变特点.
对象与方法
一、研究对象
本研究纳入2014年9月6日-2015年9月10日在上海交通大学医学院附属瑞金医院神经科门诊就诊的3个共济失调毛细血管扩张症家系共4例患者,临床诊断均符合Harding标准[4];均为汉族,男性3例,女性1例;年龄10~15岁,平均14.75岁;发病年龄14~18个月,平均15.75个月;其中例1和例2为同胞姊弟;所有患者父母(6例)均身体健康,无相似临床症状,否认近亲婚配;遗传方式符合常染色体隐性遗传.选择我院既往进行流行病学调查的200名具有相同遗传背景的正常人作为对照,均为汉族.本研究经上海交通大学医学院附属瑞金医院道德伦理委员会审核批准,所有患者及其父母均知情同意并签署知情同意书.
二、研究方法
1.样本采集 采集4例患者及其父母外周静脉血各2 ml,予乙二胺四乙酸(EDTA,国药集团化学试剂有限公司)抗凝.采用标准酚氯仿法提取患者及其父母基因组DNA,操作步骤严格按照试剂盒说明书进行.采用多功能酶标仪(美国Biotek公司)检测DNA纯度.
2.基因检测 采用全外显子测序(WES)对先证者外周静脉血DNA进行检测并进行生物信息学分析,发现存在ATM基因突变.根据测序结果采用聚合酶链反应(PCR)扩增ATM基因相应外显子编码区,再行Sanger测序验证.针对ATM基因第10、49、57、62、63外显子及侧翼序列设计引物,由英潍捷基(上海)贸易有限公司合成,正向引物序列分别为ATM⁃10F:5'⁃CTCCCAAATGCTGGGAAAC⁃3'、ATM⁃49F:5'⁃AATTTGTGTTGGGCCACATT⁃3'、ATM⁃57F:5'⁃GGGCAGGCTCTCAAACATC ⁃3'、ATM ⁃62F:5'⁃AAGTGTGCATGATGTTTGTTCC ⁃3'、ATM ⁃63F:5'⁃GACCAAGAATGCAAACGAAA⁃3',反向引物序列分别为 ATM⁃10R:5'⁃CTTCTCGGCCAAACAAGAAA⁃3'、ATM⁃49R:5'⁃CGAAAAGAACCCAAAAGACC⁃3'、ATM⁃57R:5'⁃TTTTCACTCACACACTTTCATTCT⁃3'、ATM⁃62R:5'⁃CAGTGCCTTCTTCCACTCC⁃3'、ATM⁃63R:5'⁃CTGGGATTACAGGCGTGAG⁃3'.PCR 反应体系 25 μl,依次加入 dNTPs 2.50 mmol、2XGC 缓冲液Ⅰ12.50 μl、各引物序列 5 pmol、模板 DNA 100 ng、r⁃Taq 1U,再加水补充至 25 μl;反应条件为 95 ℃预变性5 min,95℃ 90 s、62 ℃ 1 min、72℃ 1 min,共循环35次,72℃延伸10 min.然后将Sanger测序结果与人类基因组ATM基因序列(GenBank序列号:NM_000051)进行比对,采用DNAStar软件包中SeqMan软件(美国DNAStar公司)进行基因突变分析.针对未报道的基因突变,对200例具有相同遗传背景的正常对照者进行相应基因检测,并采用Mutation Taster软件[5]预测其致病性.
结 果
一、临床特征
例1女性,25岁,主因行走不稳25年、扭转性痉挛反复发作11年,于2014年9月6日至我院神经科门诊就诊.患者刚会行走(1岁)时即出现行走不稳,呈醉酒步态;16个月时出现球结膜充血;4~5岁时可见眼睛有明显血丝;5岁时出现扭转性痉挛,表现为头向两侧扭转痉挛,发作频率逐渐增加,逐渐出现四肢痉挛,头部MRI未见明显异常;6岁时头部抖动更加明显,经肌肉注射A型肉毒毒素后症状缓解;8岁时再行MRI检查显示小脑萎缩;9岁时不能独立行走,需搀扶,进行性加重;24岁时完全不能站立,困于轮椅中,同时伴言语障碍、吞咽困难和饮水呛咳,其中言语障碍自学习说话时即有体现,表现为言语含糊不清,目前言语难以理解.患者足月剖宫产,出生时3.60 kg,无产伤和窒息史,2个月会俯卧抬头,3个月会翻身,5个月可独坐片刻,7个月会爬行,14个月可独立行走但常跌倒,此后逐渐不能生活自理,需喂养,17个月时会说话;8岁时有反复低热史(37.5~38.0℃),原因不明,自行痊愈;上小学后学习成绩中等,小学六年级(11岁)后辍学,反应较同龄正常儿童稍慢;14岁时曾出现一次阴道流血,后未再出现,第二性征已发育;咀嚼稍慢,饮水呛咳.父母身体健康,无相似症状,否认近亲婚配;其弟表现出相似临床症状.门诊神经系统检查:神志清楚,言语欠清晰;双眼球结膜毛细血管扩张,双眼水平眼震,眼球各向活动可,双侧瞳孔等大、等圆,直径约3 mm,余脑神经检查未见明显异常;发作性头部向后仰,四肢不自主抖动与肌阵挛,双足内翻,尤以右侧显著;四肢肌力检查不配合,肌张力增高;不能独自站立,双侧快复轮替动作、指鼻试验差,跟⁃膝⁃胫试验和Romberg征不配合;四肢腱反射对称,病理反射未引出.实验室检查血清甲胎蛋白318.61 μg/L(<7 μg/L).头部 MRI显示小脑萎缩.
例2例1之弟,男性,15岁,因进行性行走不稳伴言语不清13年,于2014年9月6日至我院神经科门诊就诊.患儿临床症状与其姊(例1)相似,包括进行性共济失调、构音障碍和球结膜毛细血管扩张,不同之处为持续性流涎和左眼向外斜视.患儿足月剖宫产,出生时3.20 kg,无产伤和窒息史;16个月时会独立行走,但行走不稳,易跌倒,出现球结膜充血;18个月时会说话,言语含糊不清;3.50岁时仍有流涎;10岁时无法独自站立,搀扶可行走;尚可生活自理,智力发育与同龄正常儿童无差异.门诊神经系统检查:神志清楚,言语欠清晰;双眼球结膜毛细血管扩张,双眼水平眼震,左眼向外斜视,双眼各向活动充分,双侧瞳孔等大、等圆,直径约3 mm,余脑神经检查未见异常;无法独自站立,困于轮椅中,无高弓足;四肢肌力5级,肌张力正常;双侧快复轮替动作、指鼻试验、跟⁃膝⁃胫试验差;腱反射减弱,病理反射未引出.实验室检查血清甲胎蛋白水平为426.33 μg/L.头部MRI显示小脑萎缩.
例3男性,14岁,主因进行性行走不稳伴言语欠流利12年,于2014年6月3日至我院神经科门诊就诊.患儿生长发育里程碑正常,12个月会说话,言语欠流利;18个月可独立行走,呈阔基步态,易跌倒,可见足内翻畸形;行走不稳和言语障碍进行性加重;9岁时出现平衡障碍,伴四肢和躯干不自主抖动;11岁时需助行器辅助行走;目前只能依靠轮椅,言语难以理解;幼儿期反复呼吸道感染史.父母身体健康,无相似症状,否认近亲婚配.门诊神经系统检查:神志清楚,言语含糊不清;双眼球结膜明显毛细血管扩张,双眼水平眼震,眼球各向活动充分,双侧瞳孔等大、等圆,直径约3.50 mm,余脑神经检查未见异常;四肢意向性震颤,紧张时加重;四肢肌力和肌张力均正常,双侧快复轮替动作和指鼻试验差,跟⁃膝⁃胫试验和Romberg征不配合,深浅感觉和腱反射正常,病理征阴性.实验室检查血清甲胎蛋白511 μg/L.头部MRI显示小脑萎缩.
例4男性,10岁,主因进行性行走不稳9年,于2015年7月7日至我院神经科门诊就诊.患儿15个月时可独立行走,易跌倒;5岁时出现四肢不自主抖动,明显行走不稳,进行性加重;目前上小学3年级,学习成绩中等偏下;病程中可见肢体抽搐.患儿足月剖宫产,出生时3.60 kg,无产伤和窒息史;12个月时会说话.父母身体健康,无相似症状,否认近亲婚配.门诊神经系统检查:神志清楚,言语尚清晰;双眼球结膜明显毛细血管扩张,无明显眼震,眼球各向活动尚可,双侧瞳孔等大、等圆,直径约3 mm,余脑神经检查未见异常;可见共济失调步态,一字步不能;四肢肌力5级,肌张力正常;双侧快复轮替动作、指鼻试验和跟⁃膝⁃胫试验差,Romberg征阳性;双上肢腱反射减弱、双下肢正常,病理反射未引出.头部MRI显示小脑萎缩.
二、基因检测


图1 例1和例2家系图 图2 Sanger测序所见 2a 例1和例2之父存在ATM基因无义突变c.8287C>T(p.Arg2763X,箭头所示) 2b 例1和例2之母存在ATM基因无义突变c.9139C>T(p.Arg3047X,箭头所示)Figure 1 Pedigree of Case 1 and Case 2. Figure 2 Sanger sequencing finding The patient's father presented ATM gene nonsense mutation c.8287C>T(p.Arg2763X)in Case 1 and Case 2(arrow indicates,Panel 2a).The patient's mother presented ATM gene nonsense mutation c.9139C>T(p.Arg3047X)in Case 1 and Case 2(arrow indicates,Panel 2b).



图3 例3家系图 图4 Sanger测序所见 4a 例3之父存在ATM基因无义突变c.8911C>T(p.Gln2971X,箭头所示) 4b 例3之母存在ATM基因缺失突变c.7141_7151delAATGGAAAAAT(p.Asn2381GlufsX18,箭头所示)Figure 3 Pedigree of Case 3. Figure 4 Sanger sequencing finding The patient's father prensented ATM gene nonsense mutation c.8911C>T(p.Gln2971X)in Case 3(arrow indicates,Panel 4a).The patient's mother presented ATM gene deficit mutation c.7141_7151delAATGGAAAAAT(p.Asn2381GlufsX18)in Case 3(arrow indicates,Panel 4b).

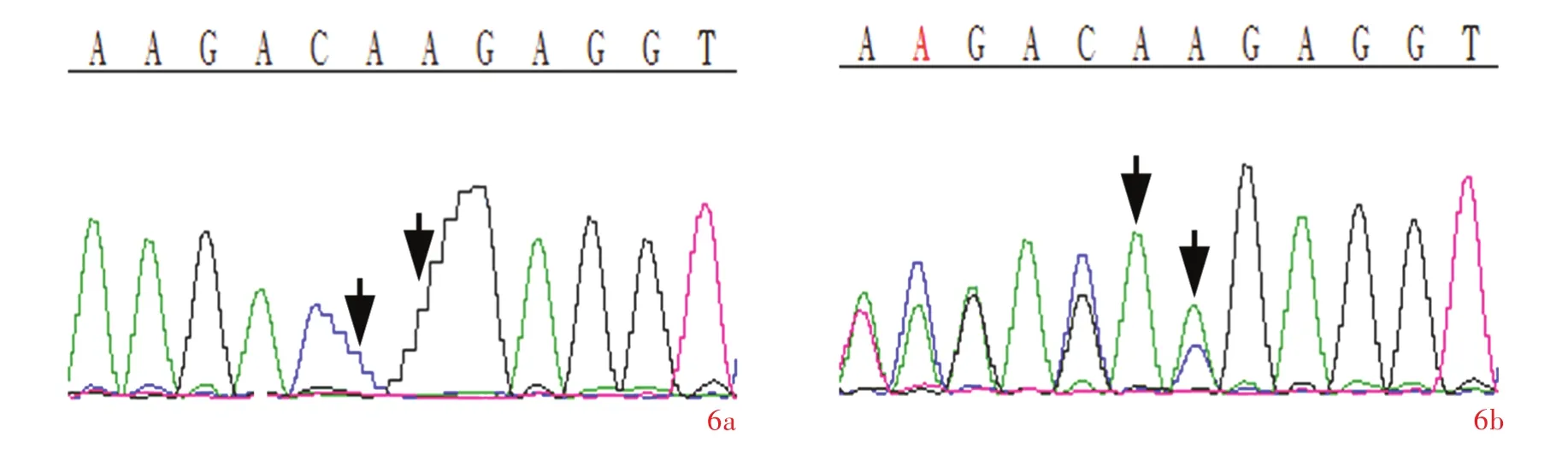
对4例患者及其父母进行ATM基因检测,共发现5种突变:例1和例2(图1)为ATM基因复合杂合突变c.8287C>T(p.Arg2763X)和c.9139C>T(p.Arg3047X),均为无义突变,其中,c.8287C>T(p.Arg2763X)突变源自其父(图2a),c.9139C>T(p.Arg3047X)突变源自其母(图2b);例3(图3)亦为ATM基因复合杂合突变,包括无义突变c.8911C>T(p.Gln2971X)和缺失突变c.7141_7151delAATGGAAAAAT(p.Asn2381GlufsX18),其中,c.8911C>T(p.Gln2971X)突变源自其父(图4a),c.7141_7151delAATGGAAAAAT (p.Asn2381GlufsX18)突变源自其母(图4b),此2种突变均未见诸报道,经Mutation Taster软件[5]预测其致病率均为1,且未在200例正常对照者中检出该突变;例4(图5)为ATM基因纯合突变c.1402_1403delAA(p.Lys468GlufsX18,图6a),其父母均为该突变携带者(图6b).
讨 论
ATM基因定位于11q22~q23,覆盖长度160 kb的基因组DNA,包含66个外显子,该基因编码丝氨酸/苏氨酸蛋白激酶(包含3056个氨基酸,相对分子质量为315X103),属磷酸肌醇3⁃激酶相关蛋白激酶(PIKK)家族成员,参与DNA损伤应答、细胞周期调控等信号转导通路,在儿童和成人各组织器官中均有表达[6].ATM基因突变具有极高的异质性,突变形式和种类具有多样性.突变形式包括错义突变、无义突变、剪切位点突变、插入或缺失突变等,其中70%为无义突变[7].突变位点遍布ATM基因全长,无明显热点突变[8].共济失调毛细血管扩张症系ATM基因纯合突变或复合杂合突变所致,其中85%系错义突变或剪切位点突变导致蛋白截短或蛋白功能缺失[8⁃9].本研究例1、例2和例3均存在ATM基因无义突变,导致编码蛋白提前终止;例4存在ATM基因纯合移码突变,改变氨基酸序列,既往文献报道该突变具有致病性[10].
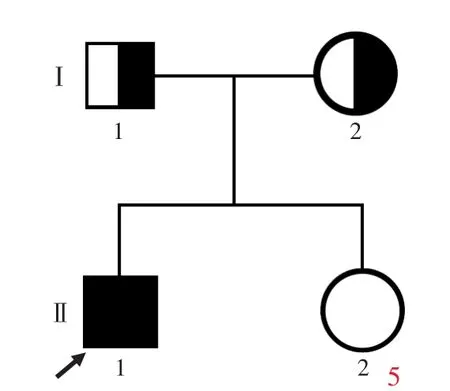

图5 例4家系图 图6 Sanger测序所见 6a 例4存在ATM基因纯合突变c.1402_1403delAA(p.Lys468GlufsX18,箭头所示) 6b 例4之父存在ATM基因杂合突变c.1402_1403delAA(p.Lys468GlufsX18,箭头所示)Figure 5 Pedigree of Case 4. Figure 6 Case 4 carried ATM gene homozygous mutation c.1402_1403delAA(p.Lys468GlufsX18, arrows indicate,Panel 6a)and the patient's fater carried heterozygous mutation c.1402_1403delAA (p.Lys468GlufsX18,arrows indicate,Panel 6b).
本研究4例患者均以步态性共济失调(2岁前)为首发症状,伴进行性加重的小脑共济失调、构音障碍,至儿童晚期(约12岁)需轮椅辅助行走,符合典型共济失调毛细血管扩张症表现[11].Moin等[12]对104例共济失调毛细血管扩张症患者进行研究,发现所有患者均伴小脑共济失调,87例(83.65%)可见球结膜毛细血管扩张、73例(70.19%)可见皮肤毛细血管扩张,78例(75.00%)伴感染,尤以肺炎、中耳炎和鼻窦炎常见.
共济失调毛细血管扩张症临床表现具有异质性,部分患者神经系统症状进展缓慢或仅轻度受累,统称为变异型共济失调毛细血管扩张症,此类患者神经系统症状较轻,头部MRI通常正常,神经系统以外受累少见,无毛细血管扩张,内分泌功能和呼吸功能正常,免疫球蛋白水平正常,生存期较长,肿瘤发生较晚[13].临床表型与ATM蛋白活性相关,ATM蛋白活性越高、临床症状越轻[13].变异型共济失调毛细血管扩张症多系ATM基因错义突变所致,与残留部分功能性ATM蛋白有关[1,13].
随着ATM基因的克隆,诸多研究显示,并非所有患者均伴小脑共济失调、球结膜和皮肤毛细血管扩张,部分患者甚至无神经系统受累表现[14⁃15].一项对成年共济失调毛细血管扩张症患者运动障碍的研究共纳入14例患者,结果显示,10/14例存在小脑共济失调,2/14例存在构音障碍和肌张力障碍,所有患者均存在运动障碍.运动障碍以肌张力障碍和皮质下肌阵挛最为常见(86%),其次为震颤(6/14)[14].Teive等[11]总结50例变异型共济失调毛细血管扩张症患者的临床表现,结果显示,86%(43/50)患者存在运动障碍,78%(39/50)存在小脑共济失调,仅16%(8/50)存在球结膜和皮肤毛细血管扩张.Verhagen等[1]总结13例变异型共济失调毛细血管扩张症患者的临床表现,发现常见神经系统症状包括小脑共济失调(12例)、肌张力障碍(10例)、舞蹈手足徐动症(9例)、静止性震颤(9例)和脊髓前角细胞性周围神经病(3例).变异型共济失调毛细血管扩张症通常于儿童期发病,毛细血管扩张可能并非其主要临床症状,共济失调进展缓慢,至成年期仅表现为轻中度共济失调,运动障碍更为常见,主要表现为肌张力障碍、肌阵挛、舞蹈样动作,帕金森样症状和震颤等.
共济失调毛细血管扩张症的早期诊断对遗传咨询具有重要意义,但以共济失调发病的患者早期诊断有一定难度,应注意与其他疾病引起的共济失调相鉴别.共济失调毛细血管扩张症是5岁前发病的最常见的常染色体隐性遗传性共济失调,Friedreich共济失调(FRDA)通常于儿童晚期发病,且伴深反射消失,Romberg征阳性和病理征阳性,可资鉴别.小脑萎缩通常在婴幼儿期不明显,至儿童晚期(约10岁)可见明显小脑萎缩.实验室检查具有一定的鉴别诊断价值,约95%共济失调毛细血管扩张症患者血清甲胎蛋白水平升高[7],核型分析可见第7号染色体与第14号染色体易位,T淋巴细胞数目减少、B淋巴细胞数目正常或仅轻度增加,IgE(80%)、IgG2(80%)和IgA(60%)明显缺陷且随疾病进展相对稳定,而IgM在病程中波动较大[7].在临床诊断基础上,结合ATM基因检测发现复合杂合突变或纯合突变有助于明确诊断.
在本研究中,例1和例2存在2种已报道的复合杂合突变,其中,c.8287C>T为无义突变,位于第57外显子,由Cavalieri等[16]于2008年率先报道,该突变可以导致1个未成熟的截短蛋白(p.Arg2763X);c.9139C>T亦为无义突变,导致蛋白羧基末端(C末端)10 个氨基酸缺失(p.Arg3047X)[17⁃18],上述2种突变位于磷酸肌醇3⁃激酶相关蛋白激酶结构域内或附近,影响磷脂酰肌醇3⁃激酶(PI3K)活性.尽管例1和例2存在相同致病性突变,但二者临床表型不尽一致,如足内翻、斜视、扭转性痉挛、肌阵挛、原发性闭经和口角流涎等,表现出疾病的遗传异质性.例3存在2种未报道的突变,包括无义突变c.8911C>T和缺失突变c.7141_7151delAATGGAAAAAT,均导致截短蛋白(p.Gln2971X,p.Asn2381GlufsX18),但这2种突变在200例正常对照者中未检出,且突变氨基酸在不同物种间高度保守,经Mutation Taster软件预测其严重影响蛋白功能,提示这2种突变均为例3的致病性突变,明确诊断尚待进一步分子功能学检测.研究显示,共济失调毛细血管扩张症患者父母均为突变基因携带者,虽无共济失调毛细血管扩张症症状与体征,但与正常对照者相比,其肿瘤易感性(乳腺癌、白血病)明显增加[19⁃20].本研究4例共济失调毛细血管扩张症患者父母目前均无肿瘤表现,尚待进一步随访研究.
本研究经基因检测明确诊断4例共济失调毛细血管扩张症患者,均为儿童期发病的进行性进展的小脑共济失调、球结膜和皮肤毛细血管扩张、免疫缺陷导致反复感染;血清甲胎蛋白水平升高;头部MRI显示小脑萎缩;同时报道2种ATM基因新突变和3种已知致病性突变,扩大ATM基因突变谱,为进一步研究奠定基础.
[1]VerhagenMM,AbdoWF,WillemsenMA,HogervorstFB,Smeets DF,Hiel JA,Brunt ER,van Rijn MA,Majoor Krakauer D,Oldenburg RA,Broeks A,Last JI,van't Veer LJ,Tijssen MA,Dubois AM,Kremer HP,Weemaes CM,Taylor AM,van Deuren M.Clinicalspectrum ofataxia⁃telangiectasia in adulthood.Neurology,2009,73:430⁃437.
[2]Gatti RA,Berkel I,Boder E,Braedt G,Charmley P,Concannon P,Ersoy F,Foroud T,Jaspers NG,Lange K,Lathrop GM,Leppert M,Nakamura Y,O'Connell P,Paterson M,Salser W,Sanal O,Silver J,Sparkes RS,Susi E,Weeks DE,Wei S,White R,Yoder F.Localization of an ataxia⁃telangiectasia gene to chromosome 11q22-23.Nature,1988,336:577⁃580.
[3]Becker⁃Catania SG,Chen G,Hwang MJ,Wang Z,Sun X,Sanal O,Bernatowska⁃Matuszkiewicz E,Chessa L,Lee EY,Gatti RA.Ataxia⁃telangiectasia:phenotype/genotype studies ofATM protein expression,mutations,and radiosensitivity.Mol Genet Metab,2000,70:122⁃133.
[4]Harding AE.The hereditary ataxiasand related disorders.Edinburgh:Churchill Livingstone,1984:325⁃326.
[5]Schwarz JM, Cooper DN, Schuelke M, Seelow D.MutationTaster2:mutation prediction for the deep⁃sequencing age.Nat Methods,2014,11:361⁃362.
[6]Chaudhary MW,Al⁃Baradie RS.Ataxia⁃telangiectasia:future prospects.Appl Clin Genet,2014,7:159⁃167.
[7]Perlman S,Becker⁃Catania S,Gatti RA.Ataxia⁃telangiectasia:diagnosis and treatment.Semin Pediatr Neurol,2003,10:173⁃182.
[8]Chun HH,GattiRA.Ataxia⁃telangiectasia,an evolving phenotype.DNA Repair,2004,3:1187⁃1196.
[9]Taylor AM, Byrd PJ. Molecular pathology of ataxia telangiectasia.J Clin Pathol,2005,58:1009⁃1015.
[10]Broeks A,de Klein A,Floore AN,Muijtjens M,Kleijer WJ,Jaspers NG,van'tVeerLJ.ATM germline mutationsin classical ataxia⁃telangiectasia patients in the Dutch population.Hum Mutat,1998,12:330⁃337.
[11]Teive HA,Moro A,Moscovich M,Arruda WO,Munhoz RP,Raskin S,Ashizawa T.Ataxia⁃telangiectasia⁃A historical review and a proposal for a new designation:ATM syndrome.J Neurol Sci,2015,355:3⁃6.
[12]Moin M,Aghamohammadi A,Kouhi A,Tavassoli S,Rezaei N,Ghaffari SR,Gharagozlou M,Movahedi M,Purpak Z,Mirsaeid Ghazi B,Mahmoudi M,Farhoudi A.Ataxia⁃telangiectasia in Iran:clinical and laboratory features of 104 patients.Pediatr Neurol,2007,37:21⁃28.
[13]Verhagen MM,Last JI,Hogervorst FB,Smeets DF,Roeleveld N,Verheijen F,Catsman⁃Berrevoets CE,Wulffraat NM,Cobben JM,Hiel J,Brunt ER,Peeters EA,Gómez Garcia EB,van der Knaap MS,Lincke CR,Laan LA,Tijssen MA,van Rijn MA,Majoor⁃Krakauer D,Visser M,van't Veer LJ,Kleijer WJ,van de Warrenburg BP,Warris A,de Groot IJ,de Groot R,Broeks A,Preijers F,Kremer BH,Weemaes CM,Taylor MA,van DeurenM,WillemsenMA.PresenceofATM proteinand residual kinase activity correlates with the phenotype in ataxia⁃telangiectasia:a genotype⁃phenotype study.Hum Mutat,2012,33:561⁃571.
[14]Méneret A,Ahmar⁃Beaugendre Y,Rieunier G,Mahlaoui N,Gaymard B,Apartis E,Tranchant C,Rivaud⁃Péchoux S,Degos B,Benyahia B,Suarez F,Maisonobe T,Koenig M,Durr A,Stern MH,Dubois d'Enghien C,Fischer A,Vidailhet M,Stoppa⁃Lyonnet D,Grabli D,Anheim M.The pleiotropic movement disorders phenotype of adult ataxia⁃telangiectasia.Neurology,2014,83:1087⁃1095.
[15]Trimis GG,Athanassaki CK,Kanariou MM,Giannoulia⁃Karantana AA.Unusual absence of neurologic symptoms in a six⁃year old girl with ataxia⁃telangiectasia.J Postgrad Med,2004,50:270⁃271.
[16]Cavalieri S,Funaro A,Pappi P,Migone N,Gatti RA,Brusco A.Large genomic mutations within the ATM gene detected by MLPA,including a duplication of 41 kb from exon 4 to 20.Ann Hum Genet,2008,72:10⁃18.
[17]Toyoshima M,Hara T,Zhang H,Yamamoto T,Akaboshi S,Nanba E,Ohno K,Hori N,Sato K,Takeshita K.Ataxia⁃telangiectasia without immunodeficiency:novel point mutations within and adjacent to the phosphatidylinositol 3⁃kinase⁃like domain.Am J Med Genet,1998,75:141⁃144.
[18]McConville CM,Stankovic T,Byrd PJ,McGuire GM,Yao QY,Lennox GG,TaylorMR.Mutationsassociated with variant phenotypes in ataxia⁃telangiectasia.Am J Hum Genet,1996,59:320⁃330.
[19]GattiRA,Tward A,Concannon P.Cancerrisk in ATM heterozygotes: a model of phenotypic and mechanistic differences between missense and truncating mutations.Mol Genet Metab,1999,68:419⁃423.
[20]Ahmed M,Rahman N.ATM and breast cancer susceptibility.Oncogene,2006,25:5906⁃5911.
Clinical phenotype and genetic characteristics of ataxia⁃telangiectasia:four cases report
ZHENG Lan1,LIU Xiao⁃li2,CAO Li21Department of Neurology,Central Hospital of Minhang District,Shanghai 201199,China
2Department of Neurology and Institute of Neurology,Ruijin Hospital,School of Medicine,Shanghai Jiaotong University,Shanghai 200025,China
CAO Li(Email:caoli2000@yeah.net)
ObjectiveTo report 4 cases of ataxia⁃telangiectasia(AT)with ATM genetic mutation and to summarize the clinical and genetic characteristics of AT by literatures review.MethodsClinical data of 4 patients from 3 AT families was collected in detail and genomic DNA of the patients and family members was extracted from peripheral blood. Whole exon sequencing(WES)and polymerase chain reaction(PCR)of Sanger sequencing was used to analyse ATM genic mutation.ResultsFour patients were characterized by progressive cerebellar ataxia with onset in childhood,oculocutaneous telangiectasia,recurrent infection caused by immunodeficiency,α⁃fetoprotein(AFP)elevation and cerebellar atrophy shown in brain MRI were presented.Sequence analysis of ATM gene revealed two known compound heterozygous mutations c.8287C>T(p.Arg2763X)and c.9139C>T(p.Arg3047X)which were nonsense mutation in Case 1 and Case 2.In Case 3,there were two compound heterozygous mutations,including nonsence mutation c.8911C>T(p.Gln2971X)and deficit mutation c.7141_7151delAATGGAAAAAT(p.Asn2381GlufsX18)both of which were not reported previously.Case 4 carried homozygotic mutation c.1402_1403delAA(p.Lys468GlufsX18).ConclusionsFour patients were diagnosed as AT with typical clinical manifestations.Patients with variant AT present mild nervous system symptom,normal head MRI and less involvement other than nervous systemt.Definite diagnosis should be dependant on ATM genetic testing.
Ataxia telangiectasia; Phenotype; Genes; Mutation
10.3969/j.issn.1672⁃6731.2017.07.008
上海市卫生和计划生育委员会科研课题(项目编号:20164Y0262);上海市闵行区中心医院课题(项目编号:2016MHJC04)
201199上海市闵行区中心医院神经内科(郑岚);200025上海交通大学医学院附属瑞金医院神经科 上海交通大学医学院神经病学研究所(刘晓黎,曹立)
曹立(Email:caoli2000@yeah.net)
This study was supported by the Scientific Research Planning of Shanghai Municipal Commission of Health and Family Planning Committee(No.20164Y0262)and the Program of Central Hospital of Minhang District,Shanghai(No.2016MHJC04).
2017⁃06⁃27)
猜你喜欢
青年文学家(2024年10期)2024-05-26 18:27:59
北京大学学报(医学版)(2022年2期)2022-11-21 03:29:12
保健与生活(2022年9期)2022-05-06 22:50:00
甘肃科技(2020年20期)2020-04-13 00:30:54
中国心血管杂志(2020年4期)2020-01-09 03:47:19
遗传(2019年5期)2019-05-21 09:58:28
岷峨诗稿(2018年2期)2018-11-14 18:51:09
医学信息(2018年5期)2018-04-20 11:03:20
家庭科学·新健康(2016年9期)2016-10-25 16:05:12
中国卫生标准管理(2015年2期)2016-01-14 03:41:39
