
费托合成铁基催化剂关键影响因素研究进展*
2017-11-13 06:24段建国王亚雄刘全生周晨亮
无机盐工业 2017年11期
段建国,王亚雄,刘全生,周晨亮
(1.内蒙古科技大学化学与化工学院,内蒙古自治区煤化工与煤炭综合利用重点实验室,内蒙古包头014010;2.内蒙古工业大学化工学院,内蒙古自治区低阶碳质资源高值功能化利用重点实验室)
综述与专论
费托合成铁基催化剂关键影响因素研究进展*
段建国1,王亚雄1,刘全生2,周晨亮1
(1.内蒙古科技大学化学与化工学院,内蒙古自治区煤化工与煤炭综合利用重点实验室,内蒙古包头014010;2.内蒙古工业大学化工学院,内蒙古自治区低阶碳质资源高值功能化利用重点实验室)
介绍了近年来费托(FT)合成铁催化剂关键影响因素取得的研究进展,围绕铁活性相物质、有效助剂、新型载体等铁基催化剂关键影响因素对铁催化剂结构和性能的影响做了综述,进一步探讨了铁活性相颗粒尺寸大小对催化剂特性的影响。最后,对未来费托催化剂的发展方向做了展望。
费托(FT)合成;铁基催化剂;活性相
费托(FT)合成是煤、天然气和生物质等碳资源间接转化成高价值的液体燃料和化工产品的一项关键技术,在环境保护和经济方面具有重要意义。在FT合成过程中,CO加氢是碳资源转化利用的重要平台。早在1923年,德国研究者即探索得到了煤经合成气催化生产液体燃料的费托催化过程[1]。之后的研究人员对催化剂和催化机理做了大量研究,并逐渐使之成为碳资源领域(气转液)转化的重要技术[2]。FT合成反应过程通常被认为是一种聚合反应,主要包括链反应、链增长和链终止等步骤。具体为采用Fe、Ru、Co等活性金属(还原态)作催化剂,CO分子和H2分子在其表面化学吸附解离进一步形成CHx(x=0~3)中间体作为聚合单体,多余的氧与氢反应生成水被移除或发生水煤气反应,CHx单体在开放的金属表面发生碳链聚合增长反应,CHx中间体生成含不同碳原子数(从一到几十)的烃类产物[3]。其主要反应方程式:
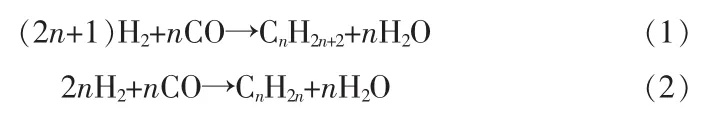
其烃类产物主要为清洁汽油、柴油、航空燃油及高价值的C2~C4低碳烯烃及芳烃等。目前,南非Sasol公司、美国美孚公司、荷兰壳牌公司等大型企业在FT合成工业化生产油品方面形成了一定的规模。同时,科研学者在FT合成技术方面的研究也取得了重大进展,但目前该技术在催化剂开发过程中仍存在很多技术难题,其中产物调控是关键问题之一。由于传统FT合成产物链增长服从聚合机理,产物选择性受Anderson-Schulz-Flory(ASF)数学模型限制,因此,如何开发可控产物选择性的FT催化剂是该技术的主要技术难题[4]。
FT催化剂活性组分以Fe、Co、Ni和Ru等为主,其金属活性顺序为Ru>Ni>Co≈Fe。其中Fe基催化剂活性较高、廉价易得、操作灵敏,是理想的FT合成催化剂,因此国内外学者多专注于对Fe基催化剂的改性研究工作中。笔者主要围绕影响FT合成催化性能的Fe基催化剂方面的关键因素,针对Fe基催化剂活性相物质、Fe活性相颗粒尺寸、有效助剂及新型碳材料载体,对其近年来取得的一些重要进展做了综述。
1 Fe基催化剂活性相物质
传统的新鲜Fe基催化剂主要以α-Fe2O3形式存在,经不同条件(气氛、压力和空速等)下还原可逐渐转变为Fe3O4、FeO、α-Fe、FexCy等物相[4-7],这些物相在催化反应过程中相互又会进一步发生转变,导致Fe物相在FT合成反应中难以确定[8]。
FT合成反应条件下,在铁基催化剂中Fe物相容易形成碳化铁,而碳化铁对CO加氢有重要作用[9-10],主要原因为在FT合成中铁原子与CO解离的碳原子显示出较高的亲和力。在FT合成中,研究者们发现了许多碳化铁的物相,如ε-Fe2C、ε′-Fe2.2C、Fe7C3、χ-Fe5C2和θ-Fe3C[4,6,10-11]。然而,大多数文献报道χ-Fe5C2为活性物相,而θ-Fe3C是失活相或是与反应无关的相[4,11],在一定操作条件下真正活性物质作用的影响仍然存疑。目前,许多研究致力于在不同的气氛、温度、压力条件下探索活性碳化铁的特征(表1),而原位表征技术对碳化铁物相特性的研究发挥显著作用。

表1 Fe基催化剂在不同条件下的碳化铁物相研究
早期研究发现,在碳化过程中,低温下易形成Fe2.2C和χ-Fe5C2;较高温度下碳化铁物相为Fe3C;更高温度下则会形成Fe7C3物相。R.J.Matyi等[16]报道了在He气氛、300℃下,Fe2.2C可以转化为Fe5C2。G.L.Caer等[17]认为碳化铁物相随反应温度的升高会引起物相发生转变。E.Boellaard等[18]发现,在反应过程中延长反应时间也会引起χ-Fe5C2向ε′-Fe2.2C转变。
最近,S.E.De等[4,11,19-20]对碳化铁的转变做了具体分析,通过将STXM与纳米管反应器相结合,发现FeKCu/SiO2催化剂在H2气氛和350℃下,Fe2O3物相向Fe3O4、Fe2SiO4和Fe0转变,而在FT反应温度为250℃、1.0 MPa及合成气作用下,Fe3O4又会进一步向Fe2SiO4和Fe0转变,随后Fe物相逐渐向碳化铁(FexCy)转变[19]。通过应用XAFS和WAXS表征技术发现,在350℃、H2气氛下,无载体的Fe基催化剂和无Cu助剂的Fe催化剂还原主要转变为α-Fe,还原的催化剂在FT反应条件0.1 MPa下易转化为θ-Fe3C,但是该催化剂由于无载体因此失活速率较快[20],表明θ-Fe3C可能有利于催化剂失活。而经CO/H2预处理后的Fe基催化剂中将出现χ-Fe2C5和γ-Fe物相,说明2种物相的存在与否与Fe基催化剂有无载体无关。经上述步骤预处理的催化剂在FT合成中展示出更高的活性和稳定性[20],说明χ-Fe2C5和γ-Fe有利于CO加氢稳定地向重碳烃类转移。同时,S.E.De等的研究小组进一步将量子化学计算与实验和理论相结合,在不同碳化气氛预处理和高压反应条件(1.0 MPa)下对Fe基催化剂从α-Fe2O3开始做了复杂的特性研究。结果发现,碳化气氛中不同的CO/H2比对Fe基催化剂中碳化物物种的影响较大。通过原位XAFS、XRD、Raman光谱等测试发现(表2),原位XRD中α-Fe2O3在350℃下碳化气氛为1%(体积分数,下同)CO/H2条件下主要形成χ-Fe5C(~56%)和θ-Fe3C(~44%),而催化剂在10%CO/H2的相同条件下,形成混合物相χ-Fe5C2、θ-Fe3C、和ε-碳物种,当20%的CO/H2和在纯CO预处理、350℃条件下,原位XRD中Fe2O3被转化为χ-Fe5C2物相(90%)和少量的ε-碳物质(10%)。
通过XRD和XAFS分析可知,在1%CO/H2气氛、350℃下显示出较高的FeCx含量。而在高压1.0 MPa、250℃下FT反应后,用1%CO/H2(合成气)预处理的催化剂,在反应过程中物相并没有大的改变。而预处理的催化剂在纯CO气氛、250℃下发生了一些变化,通过XRD和XAFS分析可知有Fe3O4形成;另一方面,在纯的CO、250℃下预处理的催化剂显示了高的CO转化率、C4+选择性和CO2活性。而经过1%的CO/H2预处理的催化剂其CO转化率和C4+的选择性较低,Raman光谱研究表明后者催化剂有石墨生成,这可能导致了其低的催化特性。因此,拥有更多金属属性的θ-Fe3C和无定型的FexC可能更有利于促进不活跃的表面含碳物种的形成速率。催化剂中Fe3O4在一定的实验条件下显示出更高的CO2形成活性,并表明Fe3O4是水煤气反应(WGS)的活性相。研究小组还从热力学方面在高压下对比高、低化学势(μc)下预处理的催化剂物相变化[20],如图1所示(表示物相转变熵在总自由能中起主要作用,表示物相转变受动力学的限制)。由图1可见,在低μc下更容易形成θ-Fe3C,高的μc下更容易形成χ-Fe5C2,而在低温200℃以下及更高的μc下ε-碳物种更易形成。K.Xu等[21]研究也发现,只有ε-Fe2C物相在低温下是稳定的,而且ε-Fe2C物相的催化剂在150℃下展示了优越的催化活性,具有较高的C5~C11选择性。
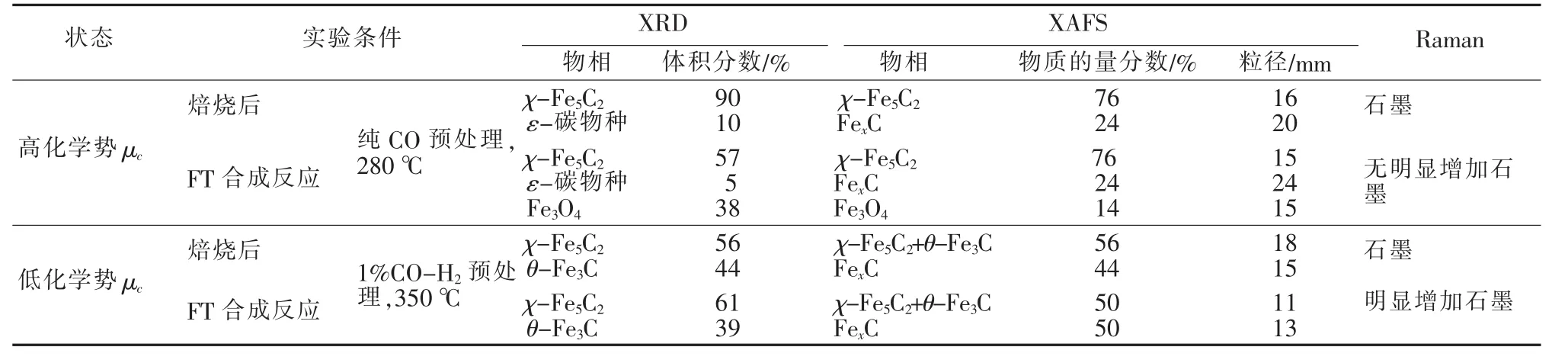
表2 Fe基催化剂(还原后Fe2O3)在不同预处理下和FT合成反应在350℃、1.0 MPa下,合成气[V(H2)/V(CO)=1]的原位特性结果[20]
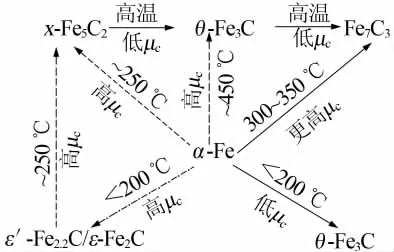
图1 碳化铁结构原子热力学研究定性解释物相转变[20]
D.B.Bukur等[22-24]在CO及合成气还原条件下对Fe基催化剂(原子比Fe∶Cu∶K=100∶0.3∶0.8)做了研究,发现CO及合成气预处理的催化剂随反应时间的增加催化活性下降,其主要原因为FT活性物相χ-Fe5C2转变为低活性氧化铁物相并增强了催化剂表面碳物种。经Mössbauer spectroscopy和XRD手段对χ-Fe5C2进行表征,发现经CO(84%~95%,体积分数)或合成气预处理后获得的χ-Fe5C2,主要转化为Fe3O4、ε′-Fe2.2C和FeCO3物相。同时在离开反应器出口的一瞬间χ-Fe5C2会被氧化为Fe3O4[25],之后它可能渗碳形成ε′-Fe2.2C,而ε′-Fe2.2C能抵制进一步的氧化。其FeCO3的形成与水煤气反应CO2产物有关。
2 Fe活性相颗粒尺寸
在FT合成反应中,活性相颗粒尺寸对于Fe基催化剂的特性研究具有重要影响,因此评估碳化铁物种的尺寸大小尤为重要。目前关于Fe基催化剂的尺寸效应的研究相对较少。早期研究指出,CO转化频率(TOF)随Ru、Fe颗粒尺寸的增大而增加[26-27]。J.Y.Park等[28]对Fe颗粒尺寸效应做了研究,通过前期合成颗粒大小在2~12 nm的γ-Fe2O3制备Fe/δ-Al2O3催化剂,在500℃下H2还原后考察不同Fe0粒径大小的催化剂。结果显示,当Fe颗粒从2.4 nm增至6.2 nm,TOF也从0.06 s-1增至0.187 s-1;而当Fe颗粒尺寸继续增至12 nm时,TOF几乎保持不变;之后随着Fe尺寸继续增大,CH4和C2~C4烃类化合物的选择性下降,C5+碳氢化合物的选择性则单独增加。近年来,一些研究学者针对颗粒尺寸对Fe基催化剂FT反应选择性的影响做了考察,并得到不同的研究结果,如表3所示。目前,关于Fe物相尺寸效应对FT合成催化产物选择性的影响还很复杂,因为在这种条件下会发生许多二级反应。
最近Z.K.Sun等[33]制备了平均粒径为22~8.3 nm的Fe2O3纳米粒子并高度分散到介孔碳纳米材料中,将碳纳米材料包含Fe作为FT合成开发的新催化剂。该催化剂随Fe粒径的增大TOF逐渐下降。随着Fe粒径从22 nm减少到8nm时,CH4选择性逐渐降低,C5+碳氢化合物选择性逐渐增加。当催化剂Fe粒径为8.3 nm时,C5+选择性达到了60%以上,并且该颗粒在没有碱金属助剂的参与下具有优越的C5+选择性,这说明Fe颗粒的大小变化导致了选择性变化的趋势。然而,这不同于J.Y.Park等的论点[28]。同时,Y.Liu等[34]用单一变量法研究了Fe颗粒大小及孔径大小对低碳烯烃的影响,通过乙二醇对FeMn/SiO2进行预处理,制备了所需的FeMn/SiO2催化剂,在300℃、1.0 MPa、V(H2)/V(CO)=1的反应条件下,通过XPS和Mössbauer spectroscopy表征结果可知,催化剂Fe/Mn原子比随Fe粒径的增大而增大,当Fe粒径从8.1 nm增至17.5 nm时,催化剂表面的Fe/Mn原子比从0.09增至0.37,而催化剂粒径大小相同时,低碳烯烃的选择性随催化剂的孔径的增大而增加,并认为在FT合成中大的孔径和小的碳化铁颗粒更有利于催化剂实现高活性和高稳定性。而不论碳化铁颗粒大小如何变化,C2~C4烃类化合物的烯烷比(O/P)随催化剂孔径由小到大的变化而明显增加,当孔径不变时,Fe或碳化铁颗粒越小越有利于形成低碳烯烃。并揭示了C2~C4烃类化合物的烯烷比对催化剂孔径变化更敏感,这主要与抑制了二级反应(加氢作用、异构化、链增长反应)形成烯烃有关。

表3 颗粒尺寸对Fe基催化剂FT反应性能的影响
3 有效助剂
在FT合成Fe基催化剂中,助剂在提升催化性能、调节产物选择性方面发挥重要作用,特别是碱金属助剂在抑制CH4选择性和提高C5+烃类化合物的选择性、CO转化低碳烯烃(C2~C4)的选择性等方面[4,9,34-35]。其主要原因为吸附在Fe表面的碱金属能增加CO的吸附热,影响铁的电子结构。特别是K能削弱CO在Fe表面的吸附能垒,导致增加CO的解离吸附[9]。C.F.Hou等[35-36]通过理论实验研究表明,K助剂通过修复晶相位有利于稳定Fe的高指数晶面和更多活性晶面。尽管在Fe表面,相对稳定性的顺序依次减弱(110)>(100)>(310)≈(211)>(321)>(210)>(111),但是DFT推算表明K的吸附能改变晶面之间的稳定性。同时,对于各指数面,高指数面拥有更低协同位的Fe原子,这些低协同Fe原子可能更有利于CO的解离,导致高的CO转化率和C5+选择性。目前,有关不同晶面间的选择性差异的文献较少,一些本质问题有待探讨。而K在影响Fe基催化剂表面CO吸附解离的同时,也影响着碳化铁物相的转变,特别是在FT合成反应中K的添加会影响Fe3O4物相向碳化铁的转变,并导致晶体发育的变化[37]。S.Li等[38-39]发现K助剂的加入有利于Fe2O3物相的还原与碳化,并减小FexC颗粒的尺寸,从而提高FT合成反应活性。
碱金属对碳化铁形成的影响在FT反应中非常重要。M.C.Ribeiro等[40]通过原位XPS表征手段研究了碱金属元素促进碳化铁形成的机理。其碳化率增加的顺序:无助剂>Li>Na>K=Rb=Cs助剂。铁物种在催化剂Fe、Si、碱性金属(Li、Na、K、Rb、Cs)原子比为100∶4.6∶5.1、在CO/He、290℃下还原10 h,碳化后产物主要为χ-Fe5C2和Fe3O4,它指出碱金属(K+、Rb+、Cs+)的添加逐渐增强CO的解离吸附率和CO吸附表面覆盖率。这可能阻止了加氢能力和烯烃再吸附的能力,有利于更高的C5+和低碳烯烃(C2~C4)选择性。W.Ngantsoue-Hoc等[41]考察了碱金属助剂K、Na、Li、Cs或Rb对Fe基催化剂在FT合成的活性与选择性的影响,结果发现K、Na助剂与Li、Cs或Rb助剂相比具有较高的CO转化率和水煤气反应活性,而Li、Cs或Rb助剂会降低Fe基催化剂加氢活性,但碱性助剂普遍能提高C2碳氢化合物的选择性。进一步研究发现,不同碱金属助剂添加会促进催化剂表面优先暴露的χ-Fe5C2晶相并有利于提高Fe3O4向碳化铁的转变速率。
Mn是另一种有效的助剂,研究认为:Mn不仅能提高Fe催化剂的还原能力,而且适量Mn助剂的添加能提高铁活性相的分散度,减小铁活性相的粒径。在FT催化反应过程中,部分氧化锰微粒会迁移并覆盖在活性组分铁颗粒的表面,形成小集团分布,降低催化剂烯烃的二次加氢及甲烷化反应,从而使催化剂的催化活性提高。近期的研究发现,Mn能增加CO转化率和降低Fe基催化剂CH4的选择性[4,42],此外还能增加C2~C4烯烃及C5+选择性[43]。但是Mn的作用机理仍然不够明朗。
近年来,A.Campos等[44]通过XANES考察了添加Mn对Cu负载的Fe/SiO2催化剂的影响,并发现Mn能替代Fe3O4中的八面体位,可能形成(Fe1-yMny)3O4,该混合氧化物相有利于形成更小类型的FexC,而FexC对于促进CO加氢有更高的活性。张敬畅等[45]采用草酸铁为铁源,椰壳炭为载体,制备得到Fe-Mn-K/AC催化剂。发现Fe-Mn-K/AC催化剂晶体结构主要以Fe嵌入MnO中形成的(Fe、Mn)O结构存在,可增大催化剂的表面积,提高铁的分散度。同时,K、Mn助剂具有协同作用,有助于提高低碳烯烃的选择性和催化活性。这说明助剂与活性相之间的相互作用和催化剂催化活性有密切联系,Mn在FT催化的活性和选择性方面发挥着积极作用。
然而,一些研究得到了不同的结果。M.C.Ribeiro等[46]发现Mn助剂添加到Fe基催化剂中会抑制Fe物相向χ-Fe5C2转变,导致Fe3O4含量增加,Mn含量的增加还会导致更高的CH4和C2~C4选择性,降低低碳烯烃选择性,促进CO选择性提高。J.D.Xu等[47]制备了Fe3-xMnxO4/CNT(x=0~0.5)纳米颗粒,发现适量Mn的添加(Mn与Fe原子比≤0.01),会提升C5+收率和C2~C4低碳烯烃的选择性,过量的Mn添加(Mn与Fe原子比>0.024)会降低催化活性,不会进一步提高C2~C4烯烃的选择性。同时,过量Mn的添加会抑制还原过程中的FeO还原到Fe,并进一步抑制Fe的碳化速率。因此,关于Mn助剂如何作用影响Fe基催化剂的FT催化活性及低碳烯烃选择性还有待进一步研究。
4 新型碳材料载体
在FT合成中催化剂活性相的载体在活性、选择性及稳定性上发挥着重要的影响。近年来,碳纳米材料如活性炭、碳纳米管(CNTs)、碳纳米纤维以及石墨烯等新型材料不断地开发,其凭借大的比表面积、可调节的孔径与孔结构及通过表面修饰改性可以调节金属前躯体与载体的相互作用等优势,常常被作为载体用于FT催化剂的研究。
一些研究已经证实在FT催化反应中CNTs对活性金属组分有积极的影响[48-49],表现在CNTs为金属纳米颗粒提供了一种有吸引力的限域环境,简称“限域效应”。W.Chen等[5,50]成功制备了位于CNT管腔内和管腔外且前驱体Fe2O3尺寸相近的铁基催化剂,并比较了CNT管腔内外铁催化剂在FT合成反应中的催化性能,发现在CNT腔内Fe2O3容易还原,而且Fe在CNT腔内(Fe-in-CNT)比在腔外(Feout-CNT)表现出更高的CO转化率和C5+选择性,CH4和C2~C4碳氢化合物的选择性更低,并进一步发现CO与CNT内表面有较强的相互作用[50]。M.M.Abbaslou等[8,51]也研究了在碳纳米管内外的Fe基催化剂,发现Fe-in-CNT催化剂形成的C5+碳氢化合物含量较高,在CNT催化剂中的Fe粒径大小保持不变,而Fe-out-CN中Fe的颗粒从5~9 nm增至6~24 nm,Fe-in-CNT中经原位XRD表征得到有较高含量的碳化铁。实验还认为,CNT催化剂具有较高的还原特性更可能是由于受CNT管内外不同的电子特性及限域效应的影响。X.Q.Chen等[52]合成了豆荚状碳纳米管封装的铁纳米粒子催化剂(Pod-Fe),比较了该催化剂酸洗前后的性能。结果表明,酸洗前后2个催化剂在不同的空速和温度反应条件下都有着良好的低碳烯烃选择性。结合反应后TEM和XRD的结果发现,碳管外部裸露的铁粒子会在反应过程中形成碳化铁物种,并随着反应进行产生聚集,同时伴有大量积碳,导致活性迅速下降;而碳层包覆的铁粒子具有高的稳定性,并且没有积碳的产生。
J.Guan等[53]制备了CNTs限域FeN纳米颗粒(FeN-in-CNT),发现在FT反应过程中,CNTs内的FeN微粒具有更高的催化活性。这种FeN催化剂显示的活性比还原性Fe催化剂或FeN/SiO2催化剂高5~7倍,同时,FeN-in-CNT比Fe-out-CNT催化剂拥有更高的活性,FeN对于CO加氢是一种更高效的活性相。主要原因为FeN-in-CN和FeN-out-CNT催化剂在FT反应中FeN转变为Fe2CxN1-x、FeCxN1-x和γ″-FeN,而且在反应过程中没有Fe3O4存在。表面碳主要通过扩散到FeN晶格中替代N原子形成FeCxN1-x,FeN-in-CNT和FeN-out-CNT催化剂具有高的水煤气变换反应(WGS)活性和高的C2~C4碳氢化合物选择性[8,54]。该小组进一步添加Mn、K对FeN-in-CNT做了性能研究,通过XRD及Mössbauer spectroscopy表征发现Mn有助于主要物相FeN向γ′-Fe2N转变,能降低CO的转化率,增加烯烃选择性。关于新型碳材料石墨烯负载铁的研究也有所报道。杨敬贺等[55]采用水热法合成多孔球形磷酸铁/氧化石墨复合物(FePO/GO),经H2还原,获得磷化铁/石墨烯纳米复合物(FeP/G)。实验发现石墨烯不但能调控FePO的结构,而且还能促进FePO的还原。同时能降低Fe电子结合能。还原后的FeP/G具有高的催化活性,其CO转化的催化活性是相同条件下FeP的70多倍,烯烷比是其3倍。经长时间反应(20 h),FeP/G催化活性没有降低,其在FT合成反应中催化结构稳定。
5 结语
FT合成反应是非石油路径制备石化产品的一个重要步骤,该反应的关键是开发研究高性能催化剂,最终实现调控目标产物的选择性。目前,FT合成反应在高价值化学品方面以煤气化经甲醇制备低碳烯烃取得了重大进展,如能实现合成气直接制备低碳烯烃将更具有竞争力和经济价值。但是目前这项技术在该领域还不成熟,仍需要解决关键技术难题,特别是开发高选择性催化剂。因此,未来的一段时间内,应将催化与先进表征、新型材料、分子模拟等科学技术密切结合,进一步深化催化剂活性相、助剂等作用机理,并通过结合原位表征技术探索有效助剂对活性相的作用机理、催化剂活性相作用及纳米催化机理等,为催化剂理论与实践研究奠定基础,并在此基础上推进新型催化剂的研发。
[1]James O O,Chowdhury B,Mesubi M A,et al.Reflections on the chemistry of the Fischer-Tropsch synthesis[J].RSC Advances,2012,2(19),7347-7366.
[2]Torres Galvis H M,de Jong K P.Catalysts for production of lower olefins from synthesis gas:A review[J].ACS Catalysis,2013,3(9):2130-2149.
[3]刘忠范.合成气定向转化低碳烯烃[J].物理化学学报,2016,32(4):803-804.
[4]De S E,Weckhuysen B M.The renaissance of iron-based Fischer-Tropsch synthesis:on the multifaceted catalyst deactivation behaviour[J].Chem.Soc.Rev.,2008,37(12):2758-2781.
[5]Chen W,Pan X L,Bao X L.Tuning of redox properties of iron and iron oxides via encapsulation within carbon nanotubes[J].J.Am.Chem.Soc.,2007,129(23):7421-7426.
[6]Chen W,Pan X,Willinger M G,et al.Facile autoreduction of iron oxide/carbonnanotu been capsulates[J].J.Am.Chem.Soc.,2006,128(10):3136-3137.
[7]王维佳,罗明生,李金林.硅对铁基费-托合成催化剂的影响[J].催化学报,2007,28(10):925-930.
[8]Abbaslou R M M,Tavasoli A,Dalai A K.Effect of pre-treatment on physico-chemicalproperties and stability of car bonnano tubes supported iron Fischer-Tropsch catalysts[J].Appl.Catal.A,2009,355(1):33-41.
[9]Zhang Q H,Kang J C,Wang Y.Development of novel catalysts for Fischer-Tropschsyn the sis:tuning the product selectivity[J].Chem.Cat.Chem.,2011,42(1):1030-1058.
[10]Ninga W S,Koizumi N,Yamadab M.Improvement of promoters on theFischer-Tropschactivity of mechani cally mixed Fecatalysts[J].Catal.Commun.,2007,8(3):275-278.
[11]De S E,Beale A W,Nikitenko S.et al.Local and long range order in promoted iron-based Fischer-Tropsch catalysts:A combined in situ X-ray absorption spectroscopy/wide angle X-ray scattering study[J].Journal of Catalysis,2009,262(2):244-256.
[12]王瑞雪,吴宝山,李永旺.单相碳化铁的制备及其表面吸附性质[J].催化学报,2012,33(5):863-869.
[13]Cano L A,Cagnoli M V,Bengoa J F,et al.Effect of the activation atmosphere on the activity of Fe catalysts supported on SBA-15 in the Fischer-Tropsch synthesis[J].J.Catal.,2011,278(2):310-320.
[14]Yang C,Zhao,H B,Hou Y.et al.Fe5C2nanoparticles:A facile bromide-inducedsynthesis and as an active phase for Fischer-Tropsch synthesis[J].J.Am.Chem.Soc.,2012,134(134):15814-21.
[15]定明月,杨勇,相宏伟,等.费托合成Fe基催化剂中铁物相与活性的关系[J].催化学报.2010,31(9):1145-1150.
[16]Matyi R J,Schwartz L H,Butt J B.Particle size distribution,and related measurements of supported metal catalysts[J].Catalysis Reviews,1987,29(1):41-99.
[17]Caer G L,Dubois J M,Pijolat M,et al.Characterization by Möessbauer spectroscopy of iron carbides formed by Fischer-Tropsch synthesis[J].J.Phys.Chem.,1982,86(24):4799-4808.
[18]Boellaard E,Kraan A V D,Geus J W.et al.Behaviour of cyanidederived CuxFe/Al2O3catalysts during Fischer-Tropsch[J].Applied Catalysis A General,1999,147(1):175-187.
[19]De S E,Swart I,Creemer J F,et al.Nanoscale chemical imaging of a working catalyst by scanning transmission X-ray microscopy[J].Nature,2008,456(7219):222-225.
[20]De S E.Cinquini F,Beale A M,et al.Stability and reactivity of εχ-θ iron carbide catalyst phases in Fischer-Tropsch synthesis:Controlling μc[J].J.Am.Chem.Soc.,2010,132(42):14928-14941.
[21]Xu K,Sun B,Lin J,et al.ε-iron carbide as a low-temperature Fischer-Tropsch synthesis catalyst[J].Nature Communications,2014,5:5783-5783.
[22]Bukur D B,Okabe K,Rosynek M P,et al.Activation studies with a precipitated iron catalyst for Fischer-Tropsch synthesis:I.Characterization studies[J].J.Catal.,1995,155(2):353-365.
[23]Bukur D B,et al.Activation studies with aprecipitated iron catalyst for Fischer-Tropsch synthesis:Ⅱ.Reaction studies[J].J.Catal.,1995,155(2):366-375.
[24]Bukur D B,Koranne M,Lang X S,et al.Pretreatment effect studies with a precipitated iron Fischer-Tropsch catalyst[J].Appl.Catal.A General,1995,126(1):85-113.
[25]Duvenhage D J,Coville N J.Deactivation of a precipitated iron Fischer-Tropsch catalyst-A pilot plant study[J].Appl.Catal.A General,2006,298(1):211-216.
[26]Kang J C,Deng W P,Zhang Q H,et al.Ru particle size effect in Ru/CNT-catalyzed Fischer-Tropsch synthesis[J].Journal of Energy Chemistry,2013,22(2):321-328.
[27]Jones V K,Neubauer L R,Bartholomew C H.Effects of crystallite size and support on the carbon monoxide hydrogenation activity/selectivity properties of iron/carbon[J].J.Phys.Chem.,2002,90(20):4832-4839.
[28]Park J Y,Lee Y J,Khanna P K,et al.Alumina-supported iron oxide nanoparticles as Fischer-Tropsch catalysts:Effect of particle size of iron oxide[J].J.Mol.Catal.A,2010,323(1/2):84-90.
[29]Cheng K,Virginie M,Ordomskya V V,et al.Pore size effects in high-temperature Fischer-Tropsch synthesis over supported iron catalysts[J].Journal of Catalysis,2015,328,139-150.
[30]Galvis H M T,Bitter J H,Davidian T,et al.Iron particle size effects for direct production of lower olefins from synthesis gas[J].J.Am.Chem.Soc.,2012,134(39):16207-16215.
[31]Barkhuizen D,Mabaso E I,Viljoen E,et al.Experimental approaches to the preparation of supported metal nanoparticles[J].Pure Appl.Chem.,2006,78(9),1759-1769.
[32]Jung H J,Walker P L,Vannice A.CO hydrogenation over well-dispersed carbon-supported iron catalysts[J].J.Catal.,1982,75(2):416-422.
[33]Sun Z K,Sun B,Qiao M H,et al.A General chelate-assisted coassembly to metallic nanoparticles incorporated ordered mesoporous carbon catalysts for Fischer-Tropsch synthesis[J].J.Am.Chem.Soc.,2012,134(42),17653-17660.
[34]Liu Y,Chen J F,Zhan Y.The effect of pore size or iron particle size on the formation of light olefins in Fischer-Tropsch synthesis[J].RSC Adv.,2015,5(37):781-788.
[35]Huo C F,Wu B S,Gao P,et al.The mechanism of potassium promoter:Enhancing the stability of active surfaces[J].Angew.Chem.Int.Ed.,2011,123(32):7541-7544.
[36]Huo C F,Li Y W,Wang J G,et al.Insight into CH4formation in iron-catalyzedFischer-Tropschsynthesis[J].J.Am.Chem.Soc.,2009,131(41):14713-14721.
[37]沈菊李.费托合成Fe1-xO基熔铁催化剂的研究[D].杭州:浙江工业大学,2005.
[38]Li S,Li A,Krishnamoorthy S,et al.Effects of Zn,Cu and K promoters on the structure and on the reduction,carburization and catalytic behavior of iron-based Fischer-Tropsch synthesis catalysts[J].Catal.Letters,2001,77(4):197-205.
[39]Li S,Meitzner G D,Iglesia E.Structure and site evolution of iron oxide catalyst precursors during the Fischer-Tropsch synthesis[J].Phys.Chem.B,2001,105(24):5743-5750.
[40]RibeiroMC,JacobsG,Davis B H,et al.Fischer-Tropsch synthesis:an in-situ TPR-EXAFS/XANES investigation of the influence of Group I alkali promoters on the local atomic and electronic structure of carburized iron/silica catalysts[J].Phys.Chem.C,2010,114(17):7895-7903.
[41]Ngantsoue-Hoc W,Zhang Y Q,O′Brien R J,et al.Fischer-Tropsch synthesis:Activity and selectivity for group I alkali promoted ironbased catalysts[J].Applied Catalysis A:General,2002,236(1/2):77-89.
[42]Li T Z,Wang H L,Yang Y,et al.Effect of manganese on the catalytic performance of an iron-manganese bimetallic catalyst for light olefin synthesis[J].Journal of Energy Chemistry,2013,22(4):624-632.
[43]Lohitham N,Goodwinjr J G.Effect of K promotion of Fe and FeMn Fischer-Tropsch synthesis catalysts:Analysis at the site level using SSITKA[J].Journal of Catalysis,2008,260(1):7-16.
[44]Campos A,Lohitharn N,Roy A,et al.An activity and XANES study of Mn-promoted,Fe-based Fischer-Tropsch catalysts[J].Appl.Catal.A,2010,375(1):12-16.
[45]张敬畅,卫国宾,曹维良.合成气制低碳烯烃用Fe/AC催化剂的制备及性能表征[J].催化学报,2003,24(4):259-264.
[46]Ribeiro M C,Jacobs G,Pendyala R,et al.Fischer-Tropsch synthesis:Influence of Mn on the carburization rates and activities of Febased catalysts by TPR-EXAFS/XANES and catalyst testing[J].J.Phys.Chem.C,2011,115(11):4783-4792.
[47]Xu J D,Zhu K T,Weng X F,et al.Carbon nanotube-supported Fe-Mn nanoparticles:A model catalyst for direct conversion of syngas to lower olefins[J].Catalysis Today,2013,215(41):86-94.
[48]Pan X L,Bao X H.Reactions over catalysts confined in carbon nanotubes[J].ChemInform,2009,47(10):6271-6281.
[49]Pan X L,Fan Z L,Chen W,et al.Enhanced ethanol production inside carbon nanotube reactors containing catalytic particles[J].Nature Materials,2007,6(7):507-511.
[50]Chen W,Fan Z L,Pan X L,et al.Effect of confinement in carbon nanotubes on the activity of Fischer-Tropsch iron catalyst[J].J.Am.Chem.Soc.,2008,130(29):9414-9419.
[51]Abbaslou R M M,Tavassoli A,Soltan J,et al.Iron catalysts supported on carbon nanotubes for Fischer-Tropsch synthesis:Effect of catalytic site position[J].Appl.Catal.A,2009,367(1/2):47-52.
[52]Chen X Q,Deng D H,Pan X L,et al.Iron catalyst encapsulated in carbon nanotubes for CO hydrogenation to light olefins[J].Chinese Journal of Catalysis,2015,36(9)1631-1637.
[53]Guan J,Pan X L,Liu X,et al.Syngas segregation induced by confinement in carbon nanotubes:A combined first-principles and montecarlostudy[J].J.Phys.Chem.C,2009,113(52):21687-21692.
[54]Pan X L,Fan Z L,Chen W,et al.Enhanced ethanol production inside carbon nanotube reactors containing catalytic particles[J].Nature Materials,2007,6(7):507-511.
[55]杨敬贺,赵博,赵华博,等.磷化铁/石墨烯纳米复合物的制备及其在F-T合成反应中的应用[J].化学学报,2013,71(10):1365-1368.
Research advance in key influencing factors of iron-based catalyst for Fischer-Tropsch synthesis
Duan Jianguo1,Wang Yaxiong1,Liu Quansheng2,Zhou Chenliang1
(1.Inner Mongolia Key Laboratory of Coal Chemical Industry and Comprehensive Utilization,School of Chemistry and Chemical Engineering,Inner Mongolia University of Science&Technology,Baotou 014010,China;2.Inner Mongolia Key Laboratory of High-Value Function Utilization of Low Rank Carbon Resource,School of Chemical Engineering,Inner Mongolia University of Technology)
The recent researching advance of the key influencing factors of iron-based catalyst for Fischer-Tropsch(FT)synthesis was summarized.The influences of key factors,including the active phases,the effective promoters,and the new carriers of iron-based catalysts,on the structure and performance of the iron-based catalysts were reviewed.The future development direction of FT catalyst was also prospected.
Fischer-Tropsch(FT)synthesis;iron-based catalyst;active phase
TQ138.11
A
1006-4990(2017)11-0001-07
内蒙古自治区自然科学基金项目(2015BS0206)、内蒙古科技大学创新基金项目(2015QDL09)、内蒙古褐煤热解提质和综合梯级利用过程中若干关键技术的基础研究。
2017-05-26
段建国(1989—),男,硕士研究生,主要从事煤基能源催化剂的研制。
周晨亮
联系方式:627063044@qq.com
猜你喜欢
农业研究与应用(2021年2期)2021-08-12
石油炼制与化工(2021年3期)2021-03-23
今日农业(2020年20期)2020-11-26
世界有色金属(2020年4期)2020-05-16
化工时刊(2020年11期)2020-01-12
中国特种设备安全(2019年9期)2019-12-03
橡胶工业(2015年8期)2015-07-29
中国洗涤用品工业(2015年9期)2015-02-28
中国洗涤用品工业(2015年5期)2015-02-28
中国工程咨询(2015年2期)2015-02-14
