
遗传密码子扩充系统表达载体及其稳定细胞系的构建
2017-11-06 01:24杨丹高鹏李玉霞凌焱陈惠鹏
生物技术通讯 2017年4期
杨丹,高鹏,李玉霞,凌焱,陈惠鹏
1.军事医学科学院 科技部,北京 100850;2.军事医学科学院 卫生勤务与医学情报研究所,北京 100850
遗传密码子扩充系统表达载体及其稳定细胞系的构建
杨丹1,高鹏1,李玉霞2,凌焱1,陈惠鹏1
1.军事医学科学院 科技部,北京 100850;2.军事医学科学院 卫生勤务与医学情报研究所,北京 100850
目的:构建异源表达遗传密码子扩充系统的HeLa细胞系。方法:用PCR方法扩增MmBocRS、PyltRNA基因,定向克隆到Lenti-EF1α-Puro载体中,构建Lenti-EF1α-BocRS、Lenti-PyltRNA表达质粒;在HEK293T细胞中进行病毒包装,用获得的慢病毒感染HeLa细胞,获得BocRS-tRNACUA稳定细胞株;分别应用实时定量PCR和Western印迹鉴定该稳定细胞系中MmBocRS和PyltRNA的表达;利用转染EGFP的琥珀突变体验证BocRS-tRNACUA稳定细胞株的功能。结果:酶切及测序表明Lenti-EF1α-BocRS和Lenti-PyltRNA质粒构建正确;实时定量PCR结果显示BocRS-tRNACUA稳定细胞株中PyltRNA的表达量显著提高;Western印迹结果显示BocRS-tRNACUA稳定细胞株中MmBocRS基因的表达量明显高于空白对照;转染EGFP琥珀突变体结果表明,可以通过非天然氨基酸来控制EGFP的表达。结论:构建了能稳定表达MmBocRS和PyltRNA的BocRS-tRNACUA细胞株,并实现了通过非天然氨基酸控制蛋白表达,为研究定点插入非天然氨基酸提供了细胞模型。
遗传密码子扩充;非天然氨基酸;慢病毒载体;稳定细胞株
遗传密码子扩充技术是利用正交性氨酰tRNA合成酶-tRNA(tRNA/aaRS pairs)识别mRNA上的无义密码子,并在其相应位点插入非天然氨基酸[1]。一些tRNA/aaRS pairs经过演变已能编码100多种非天然氨基酸[2],这些非天然氨基酸通常都具有特殊的物理、化学或生物特性,因此实现非天然氨基酸插入的遗传密码子扩充技术已在蛋白质研究中展现出丰富多样的应用潜质。如控制蛋白交联反应,Chin等[3]在钙调蛋白上分别引入含有叠氮基团和炔烃的非天然氨基酸,利用click reaction形成一个交联三唑结构。与传统蛋白环化形成重组蛋白相比,遗传密码子扩充技术可以在蛋白的任意位点实现,不再仅限于末端;遗传密码子扩充可以通过非天然氨基酸在蛋白的特定位点标记任意标签[4],这些标签可以是荧光探针[5]、核磁共振探针[6]、红外线探针[7],甚至可以是一些细胞毒素类或生物素类的分子[5],遗传密码子扩充的优势在于它可以把对蛋白结构的干扰降到最小,以避免影响蛋白的功能和定位。
为了方便遗传密码子扩充技术相关实验的进行和探究,我们利用慢病毒载体构建表达遗传密码子扩充系统,并进一步得到了能够实现定点插入非天然氨基酸Boc-lysine的稳定细胞株BocRS-tRNACUA,为遗传密码子扩充的应用搭建了一个平台,为遗传密码子扩充实验的开展提供了便利,也保证了实验的时空一致性。
1 材料和方法
1.1 材料
人胚肾细胞293T和人宫颈癌细胞HeLa(均用含10%胎牛血清和1%青、链霉素的DMEM培养基在37℃、5%CO2培养箱中培养,2d传代一次),质粒 Lenti-EF1α-Puro、pGP、pVSVG 和 pEG⁃FP-N1为本实验室保存;大肠杆菌DH5α感受态菌株、胶回收试剂盒、细胞RNA提取试剂盒及Quant One Step qRT-PCR(SYBR Green)试剂盒购自天根生化科技有限公司;Q5超保真DNA聚合酶、限制性内切酶BstBⅠ、BamHⅠ、ClaⅠ、XbaⅠ及T4DNA连接酶购自NEB公司;无内毒素质粒小提试剂盒、Anti Flag-Tag Mouse Monoclonal Antibody、AntiGAPDH MouseMonoclonalAnti⁃body和 Goat Anti-Mouse IgG(H+L) HRP Conju⁃gate购自康为世纪生物科技有限公司;胎牛血清、细胞培养基DMEM、OPTI-MEM、胰蛋白酶、青霉素、链霉素均购自Gibco公司;TurboFect转染试剂购自Thermo公司;非天然氨基酸Boc-lysine购自Bachem公司;MmBocRS(为方便后续检测,在MmBocRS 5'端连有Flag)、PyltRNA基因序列来源于古生菌Methanosarcina mazei,由北京奥科鼎盛生物科技有限公司合成。
1.2 质粒载体构建及鉴定
PCR扩增MmBocRS片段,正向引物为5'-TG ATTCGAATTCGCCGCCACCATGGATAAAAAAC-3',反向引物为5'-GCGGGATCCTTACAGGTTGGTAG-3'。将回收的PCR产物和质粒Lenti-EF1α-Puro分别用BstBⅠ、BamHⅠ酶切,酶切产物经1%琼脂糖凝胶电泳检测,用DNA胶回收试剂盒纯化回收目的产物,再用T4DNA聚合酶连接构成重组质粒Lenti-EF1α-BocRS,转化大肠杆菌DH5α,挑取阳性克隆培养后提取质粒,送北京奥科鼎盛生物科技有限公司测序。
同理构建重组质粒Lenti-PyltRNA。PCR扩增PyltRNA片段,正向引物为5'-CCCATCGATGATT CTTCATGCAATT-3',反向引物为5'-CTAGTCTAG ACAAGGCTTGACCGAC-3'。用 ClaⅠ、XbaⅠ分别对回收的PCR产物和质粒Lenti-EF1α-EGFP进行双酶切。
1.3 慢病毒包装
消化293T细胞制成单细胞悬液,根据实验需要将适量细胞铺至100mm皿中,待细胞密度达到 60%~80%时进行转染。将0.86μg VSVG、1.72 μgGP、3.44 μg Lenti-EF1α-BocRS、12μL 100μmol/L的PEI一起加入1.33mL PBS中形成转染复合物,将转染复合物轻微吹打混匀后室温孵育10min,直接加入铺种的平皿中,轻轻摇动平皿,使转染复合物均匀分布,将培养板置于 37℃、5%CO2恒温孵箱培养,6~8h后更换为含5%胎牛血清的DMEM培养基。分别于转染后48和72h收集平皿中的培养上清,4℃、1200r/min离心10min除去细胞碎片,用0.45μm滤器过滤,过滤液再用Amicon Ultra-15 50K于4℃、5000r/min离心20min,得慢病毒液Lenti-EF1α-BocRS,分装后于-80℃保存或立即使用。同样步骤得到慢病毒液Lenti-PyltRNA。
1.4 慢病毒液感染HeLa细胞系
感染前1d将HeLa细胞铺至12孔板,当细胞密度达80%左右时,取慢病毒液Lenti-EF1α-BocRS和Lenti-PyltRNA共同感染HeLa细胞。感染时,在含有病毒的培养上清中加入聚凝胺(polybrene)至终浓度为6μg/mL以提高病毒感染效率。感染24h后,弃去感染孔中培养基,换上500μL新鲜的完全培养基;感染48h时进行药筛,把孔内培养基换为含嘌呤霉素(终浓度为3μg/mL)的培养基进行筛选,将感染并筛选后的细胞传代,并继续施加嘌呤霉素进行维持性筛选培养;连续筛选并传3代后,冻存保留BocRS-tRNACUA稳定细胞株。
1.5 实时定量PCR检测PyltRNA的表达
从细胞中提取RNA,定量,取10ng RNA为模板,用实时定量PCR仪检测BocRS-tRNACUA稳定细胞株中PyltRNA的表达。PCR上游引物为5'-T CATGTAGATCGAACGGAC-3',下游引物为5'-GA ATCTAACCCGGCTGAA-3'。以GAPDH基因为内参,上游引物为5'-TGACTTCAACAGCGACACCC A-3',下游引物为5'-CACCCTGTTGCTGTAGCCA AA-3'。用 2-ΔΔCt分析基因的相对表达量。
1.6 Western印迹检测MmBocRS的表达
弃掉细胞里的培养基,用PBS洗一次,弃掉,用裂解液裂解,冰浴30min,12 000r/min离心10min,取上清与6×上样缓冲液混匀,煮沸5min。蛋白经10%SDS-PAGE分离后电转移至NC膜,用5%脱脂牛奶封闭液封闭1h;加 入一 抗AntiFlag-Tag Mouse MonoclonalAntibody(1∶2000)、Anti GAPDH Mouse Monoclonal Antibody(1∶2000),室温温育2h或4℃温育过夜;用TBST洗膜3次,每次10min;加入相应的二抗Goat An⁃ti-Mouse IgG(H+L) HRP Conjugate,室温温育 1h;用TBST洗膜3次,每次10min;ECL显影。
1.7 质粒转染稳定细胞株
将pEGFP-N1质粒上39Tyr突变为琥珀密码子TAG,正向引物为5'-GCGATGCCACCTAGGGCAA GCTGAC-3',反向引物为5'-GTCAGCTTGCCCTA GGTGGCATCGC-3',得到突变体质粒pEGFP-N1-UAG。转染前1d将细胞BocRS-tRNACUA铺至12孔板,当细胞密度达80%左右时进行转染。将1μg pEGFP-N1-UAG与2μL TurboFect一起加入100μL OPTI-MEM中形成转染复合物,轻微吹打混匀后室温孵育10min,直接加入铺种的平板中,轻轻摇动平板,使转染复合物均匀分布,将培养板置于37℃、5%CO2恒温孵箱中培养,6h后更换为含10%胎牛血清的DMEM培养基,其中一个孔含1mmol/L Boc-lysine。
2 结果
2.1 重组慢病毒载体的构建
从连接产物转化的平板上挑取重组慢病毒载体的单克隆,培养后提取质粒酶切,酶切产物经琼脂糖凝胶电泳鉴定(图1),与预期的全长基因大小吻合,进一步测序表明重组慢病毒载体Lenti-EF1α-BocRS和Lenti-PyltRNA构建成功。
2.2 实时定量PCR检测稳定细胞株中PyltRNA的表达
利用实时定量PCR检测BocRS-tRNACUA稳定细胞株中PyltRNA的表达量。表1结果显示,与HeLa细胞相比,BocRS-tRNACUA稳定细胞的Pyl⁃tRNA表达量显著提高。说明稳定细胞株可以稳定提供遗传密码子扩充系统中的正交性tRNA。
2.3 Western印迹检测稳定细胞株中BocRS的表达
将BocRS-tRNACUA稳定细胞和HeLa细胞分别加入适量裂解液提取蛋白,用上清液进行SDSPAGE,Western印迹检测 BocRS(BocRS 5'端连有Flag)的表达(图2),结果显示2组细胞在GAPDH大小处均出现了明显条带,稳定细胞株用Flag抗体能检测到明显的特异条带,而对照HeLa细胞无条带。说明构建的稳定细胞株能稳定表达遗传密码子扩充系统中的正交性氨酰tRNA合成酶。
2.4 验证稳定细胞株插入非天然氨基酸的功能
为检测构建的BocRS-tRNACUA稳定细胞株是否能通过添加非天然氨基酸来控制目的蛋白的表达,将pEGFP-N1的39Tyr突变为琥珀密码子TAG,测序结果如图3。将pEGFP-N1的琥珀突变体转染BocRS-tRNACUA稳定细胞株,6h后将一孔内的培养基换为含1mmol/L Boc-lysine的10%胎牛血清DMEM,另外一孔换为新鲜的10%胎牛血清DMEM,转染24h后镜下观察结果。由图4可知,只有在Boc-lysine存在的情况下pEGFP-N1的琥珀突变体才能表达绿色荧光蛋白,表明构建的BocRS-tRNACUA稳定细胞株确实可以实现遗传密码子扩充。
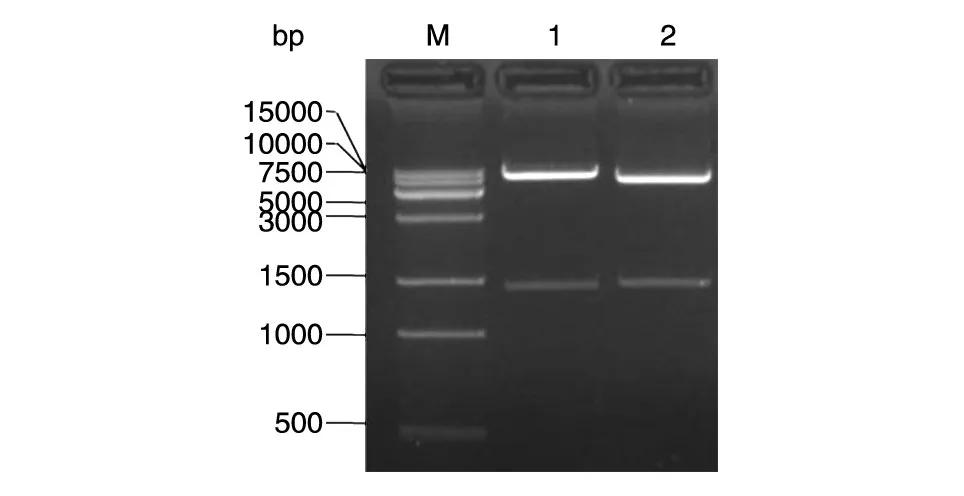
图1 重组质粒的双酶切鉴定
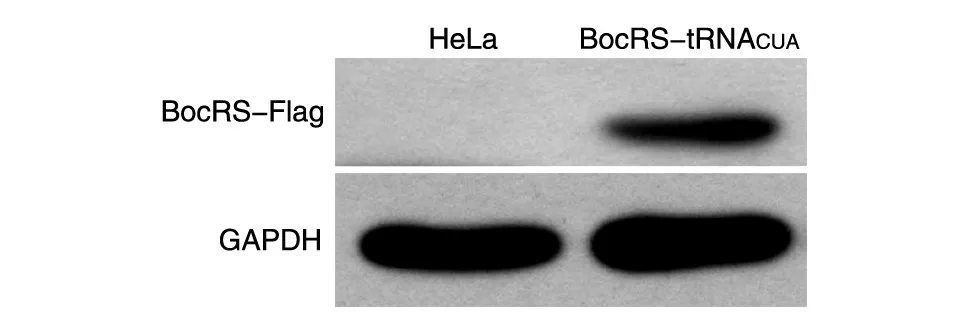
图2 Western印迹检测BocRS的表达
3 讨论
正交性氨酰tRNA合成酶-tRNA对于实现遗传密码子扩充是必不可少的,通常都是利用瞬时转染技术把它们转染到细胞内。但瞬时转染对于某些特定细胞的递送效率很低甚至不能实现,而且每次转染受细胞状态、细胞密度及实验操作者经验的影响较大,不利于保证每次试验的时空一致性。因此,发展出了病毒载体来构建遗传密码子扩充系统。使用病毒载体不仅能在常规的细胞系中实现遗传密码子扩充,甚至能在神经元细胞、胚胎干细胞及胚胎成纤维细胞中实现遗传密码子扩充。Schultz[8]等用杆状病毒表达载体实现了利用遗传密码子扩充系统插入非天然氨基酸,虽然杆状病毒具有大的基因容载量,但慢病毒载体更常见,应用更为广泛。
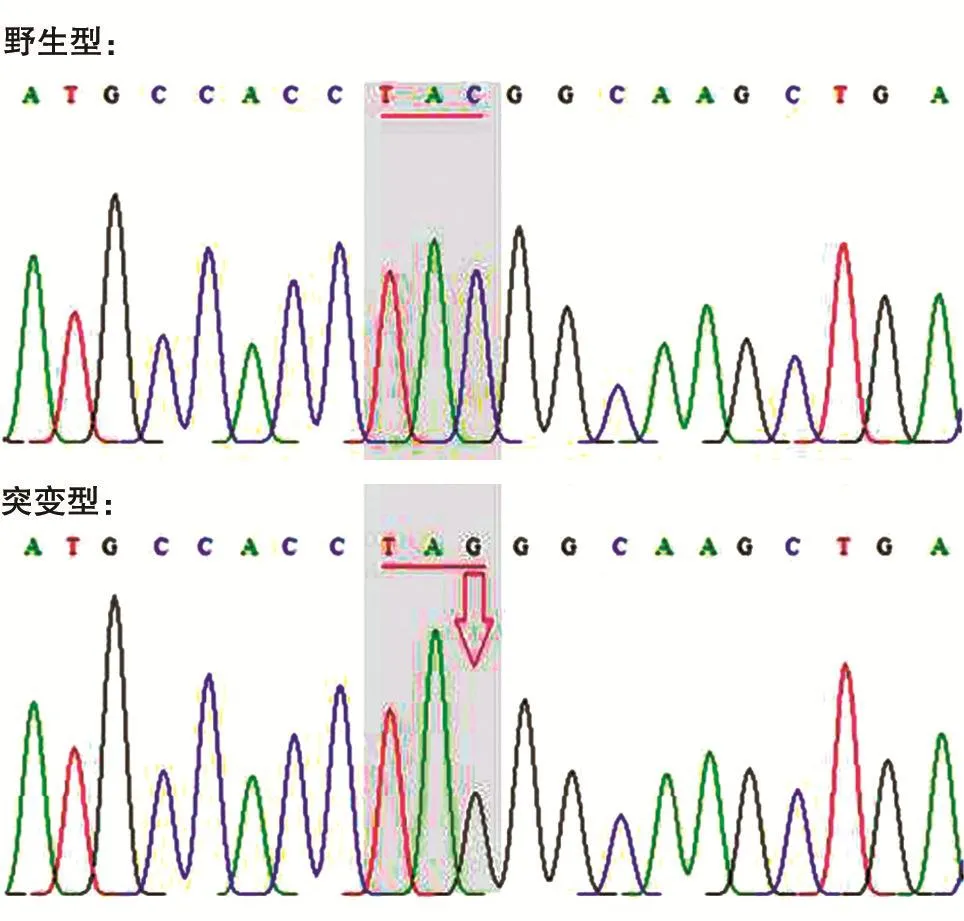
图3 pEGFP-N1琥珀突变体测序结果
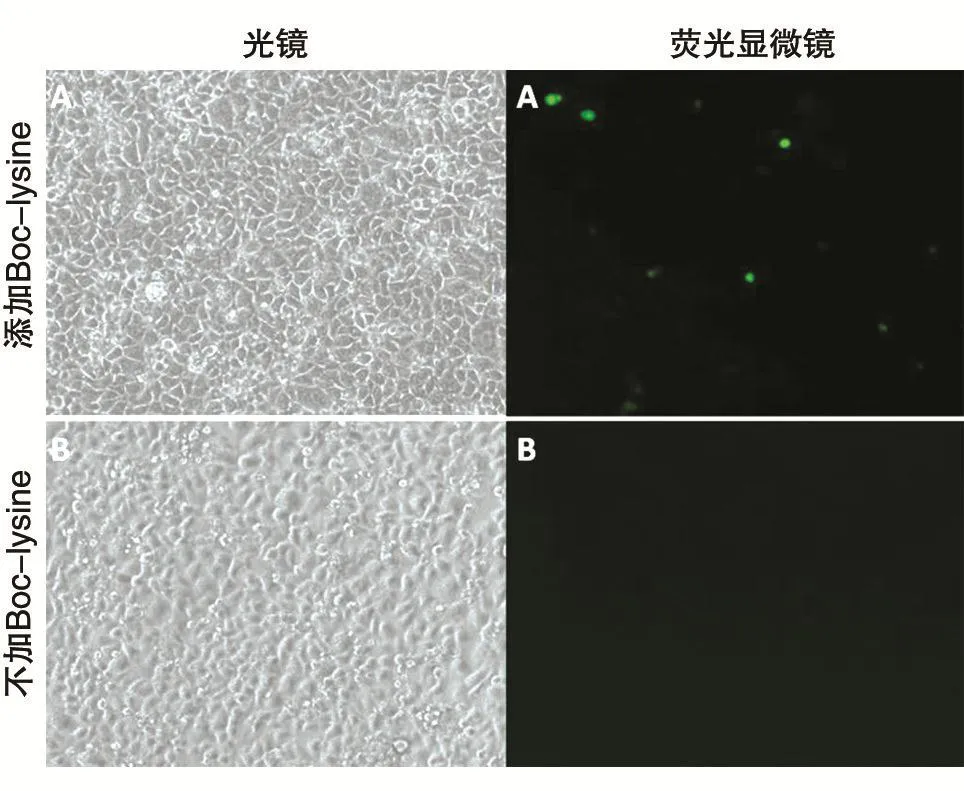
图4 BocRS-tRNACUA稳定细胞株转染结果
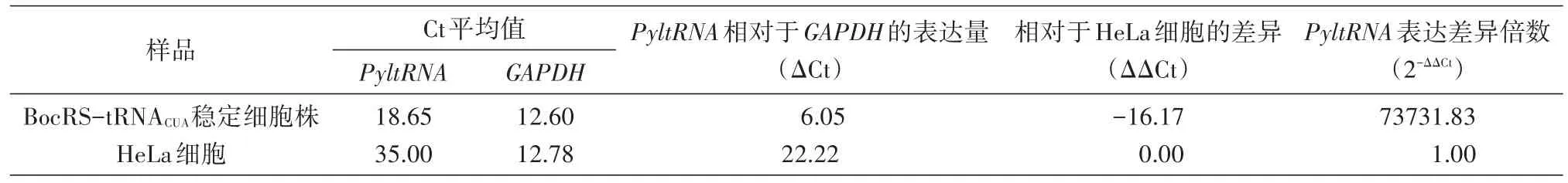
表1 实时定量PCR检测稳定细胞株和HeLa细胞中PyltRNA表达水平
遗传密码子扩充技术在病毒领域也得到了广泛应用,如Guo[9]探索了在HIV基因组中用遗传密码扩充技术制新型疫苗;Chen和Li[10]通过在丁肝病毒表面插入非天然氨基酸实现了荧光分子标记和示踪等。由于病毒的特殊性,不同病毒的生活周期差异很大,如果采用瞬时转染技术,不好控制正交性氨酰tRNA合成酶-tRNA的表达和病毒扩增之间的时间关系,而我们构建的BocRS-tRNACUA稳定细胞株可以避免这个问题。
由实验结果可知BocRS-tRNACUA稳定细胞株能稳定表达正交性MmBocRS及正交性Pyl⁃tRNACUA,并且该稳定细胞株为Boc-lysine依赖,符合遗传密码子扩充系统的要求。综上所述,我们构建的BocRS-tRNACUA稳定细胞株可以作为遗传密码子扩充系统应用的一个工具细胞,为遗传密码子技术的开展提供了便利。
[1] Chen P R,Dan G,Guo J,et al.A facile system for encoding unnatural amino acids in mammalian cells[J].Angew Chem Int Ed Engl,2009,48(22):4052-4055.
[2] Schmied W H,Elsässer S J,Uttamapinant C,et al.Efficient multisite unnatural amino acid incorporation in mammalian cells via optimized pyrrolysyl tRNA syn⁃thetase/tRNA expression and engineered eRF1[J].J Am Chem Soc,2014,136(44):15577-15583.
[3]Neumann H,Wang K,Davis L,et al.Encoding multi⁃ple unnatural amino acids via evolution of a quadru⁃plet-decoding ribosome[J].2010,464(7287):441-444.
[4] Davis L,Chin J W.Designer proteins:applications of genetic code expansion in cellbiology[J].NatRev Mol Cell Biol,2012,13(3):168-182.
[5]Wang J,Xie J,Schultz P G.A genetically encoded fluorescent amino acid[J].Proc Natl Acad Sci USA,2006,128(27):8738-8739.
[6]Deiters A,Geierstanger B H,Schultz P G.Site-specif⁃icin vivo labelingofproteinsforNMR studies[J].Chembiochem,2005,6(1):55-58.
[7] Schultz K C,Supekova L,Ryu Y,et al.A genetically encoded infrared probe[J].J Am Chem Soc,2006,128(43):13984-13985.
[8] Chatterjee A,Xiao H,Bollong M,et al.Efficient viral delivery system for unnatural amino acid mutagenesis in mammalian cells[J].Proc Natl Acad Sci USA,2013,110(29):11803-11808.
[9] Wang N,Li Y,Niu W,et al.Construction of a liveattenuated HIV-1 vaccine through genetic code expan⁃sion[J].Angew Chem Int Ed Engl,2014,53(19):4867-4871.
[10]Lin Shixian,Yan Huan,Li L,et al.Site-specific engi⁃neering of chemical functionalities on the surface of live hepatitis D virus[J].Angew Chem Int Ed Engl,2013,52(52):13970-13974.
Construction of Genetic Codon Expansion Expression Vec⁃tor and Stably Transfected Cells
YANG Dan1,GAO Peng1,LI Yu-Xia2,LING Yan1*,CHEN Hui-Peng1*
1.Department of Science and Technology,Academy of Military Medical Sciences,Beijing 100850;2.Institute of Health Service and Medical Information,Academy of Military Medical Sciences,Beijing 100850;China
*Co-corresponding authors,CHEN Hui-Peng,E-mail:chenhp0909@163.com;LING Yan,E-mail:lingyanbeijing@163.com
Objective:To establish a HeLa cell line heterologous-expressing orthogonal aminoacyl-tRNA synthe⁃tase/transfer RNA(tRNA) pairs.Methods:MmBocRS and PyltRNA genes were amplified by PCR and cloned into Lenti-EF1α-Puro vector to construct Lenti-EF1α-BocRS and Lenti-PyltRNA expression plasmids.Virus packaging was carried out in HEK293T cells.HeLa cells were infected with the obtained lentivirus to obtain BocRS-tRNACUAstably transfected cells.The expression of MmBocRS and PyltRNA in the stably transfected cells were identified by real-time quantitative PCR and Western blot respectively.The function of BocRS-tRNACUAstably transfected cells was examined by transfected amber mutant of EGFP.Results:Lenti-EF1α-BocRS and Lenti-PyltRNA plas⁃mids were constructed correctly.The results of RT-PCR showed that the expression of PyltRNA in BocRS-tRNACUAstably transfected cells was enhanced.Western blotting showed that the expression of MmBocRS gene in the stably transfected cells was significantly higher than in the blank control group.Transfection of EGFP amber mutant revealed that EGFP expression was controlled by unnatural amino acids.Conclusion:The BocRS-tRNACUAcell line stably expressing MmBocRS and PyltRNA was constructed and the expression of the protein was con⁃trolled by unnatural amino acid.It provided a cellular model for studying the insertion of non-natural amino acids.
genetic codon expansion;unnatural amino acids;lentivirus vector;stably transfected cells
Q78
A
1009-0002(2017)04-0410-05
2017-01-24
两用生物技术威胁评估(2016YFC1202400)
杨丹(1992- ),女,硕士研究生,(E-mail)science112233@163.com
陈惠鹏,(E-mail)chenhp0909@163.com;凌焱,(E-mail)lingyanbeijing@163.com
10.3969/j.issn.1009-0002.2017.04.002
猜你喜欢
生物学通报(2020年11期)2020-10-22
中成药(2018年7期)2018-08-04
食品科学(2018年10期)2018-05-23
西南医科大学学报(2015年1期)2015-08-22
医学研究杂志(2015年11期)2015-06-10
中国当代医药(2015年16期)2015-03-01
中国当代医药(2015年9期)2015-03-01
中国医药导报(2015年27期)2015-02-28
西南军医(2015年6期)2015-01-23
癌变·畸变·突变(2014年2期)2014-03-01
