
基于microRNA-mRNA配对表达谱进行联合分析的方法学进展
2017-11-06 01:24:56赵路阳张康顾成磊叶明侠范文生韩为东孟元光
生物技术通讯 2017年4期
赵路阳,张康,顾成磊,3,叶明侠,范文生,韩为东,孟元光
1.解放军总医院 a.妇产科;b.基础研究所,北京 100853;2.北京华信医院 妇产科,北京 100016;3.解放军第309医院 妇产科,北京 100091
基于microRNA-mRNA配对表达谱进行联合分析的方法学进展
赵路阳1a,张康2,顾成磊1a,3,叶明侠1a,范文生1a,韩为东1b,孟元光1a
1.解放军总医院 a.妇产科;b.基础研究所,北京 100853;2.北京华信医院 妇产科,北京 100016;3.解放军第309医院 妇产科,北京 100091
microRNA(miRNA)是一类内源性非编码小RNA,主要通过降解靶基因或抑制靶基因的翻译而调控表达。由于其作用机制复杂,目前尚未发现高效而低成本的靶标定位方法。近年来,基于碱基互补配对原理的计算机预测法被广为应用,但此方法假阳性高,不同算法所得结果差异大,会误导和干扰下游的功能学实验。因此,有研究者提出结合样本配对mRNA表达量来进一步定位靶基因,明确miRNA-mRNA相互作用方式,这种联合分析的方法受到了普遍认可。本文回顾了近年来基于miRNA-mRNA配对表达谱进行联合分析的方法学进展,并简要分析各类方法的应用范围和优缺点,为后续研究者选择方法提供参考。
microRNA;microRNA靶标预测;microRNA-mRNA相互作用;联合分析
microRNA(miRNA)是一类长19~22 nt的内源性非编码小RNA,主要在翻译水平负调控靶基因的表达[1]。绝大多数miRNA的编码基因首先在RNA聚合酶Ⅱ的作用下形成初级茎环结构,随后在Drosha的作用下形成发夹状的前体miRNA,再由Dicer剪切成为双链miRNA并整合入RNA诱导沉默复合体中。miRNA的5'端含有一段种子序列,可与mRNA的3'非翻译区(UTR)的调控元件相互作用,如果二者完全匹配,则靶mRNA被复合体降解,若为部分匹配则抑制靶mRNA的翻译。既往研究发现miRNA参与调控机体多种病理生理学过程,如细胞发育、分化、凋亡,能量代谢,肿瘤发生,心血管疾病,糖尿病等[2]。因此,全面准确地认识miRNA的作用机制并了解相关生物学功能,将有助于揭示疾病的病因,发现诊断、预后标志物,并为靶向治疗提供参考和依据。
目前,使用基于碱基互补配对原理的计算机软件对miRNA靶基因进行预测的方法因成本低,对先验知识要求少而广为应用[3]。尽管通过软件预测可以迅速获得结果,但该方法的假阳性率和假阴性率高,不同软件的结果差异较大,对下游功能学实验造成了干扰和误导[4]。究其原因,主要是miRNA与靶基因的结合位点短,特异性低,不同算法间极小的差异都会引起结果的巨大变化,同时软件预测不能考虑某些情形下特异性miRNA和mRNA表达的情况[5]。在此背景下,通过计算机辅助算法整合miRNA和mRNA配对表达谱来确定miRNA-mRNA相互作用(miRNA-mRNA interactions,MMIs)的方法应运而生。由于miRNA表达发生改变时,其所调控的mRNA表达谱也随之出现相应的表达改变,所以利用miRNA与mRNA的配对表达谱可以高精度地分辩出功能性miRNA的靶向关系。在此,我们回顾了近年来miRNA-mRNA联合分析的方法学进展,并简要分析各类方法的应用范围和优缺点,希望能为后续MMIs研究者选择分析方法提供参考。
1 MMIs分析流程
基于表达谱的MMIs分析主要包括数据的预处理、计算机分析和结果验证三部分(图1)。从数据库或高通量等方法获得的miRNA和mRNA表达量经过数据标准化、差异表达分析等预处理后,再根据实验目的、标本数量等条件选择合适的计算机分析方法进行统计学计算,获得阳性的MMIs后采用RT-qPCR、干扰试验、萤光素酶等实验进一步加以验证。本文重点对计算机分析这一步骤进行综述。
2 MMIs常见分析方法
基于配对表达谱的MMIs联合分析根据统计方法的差异可分为以下6类:相关性分析法、回归分析法、贝叶斯推理法、因果推论法、其他方法和综合分析法(表1)。由于每种方法的侧重点不同,各有优势,所以研究人员应根据数据类型和研究目选择最佳的分析方法。
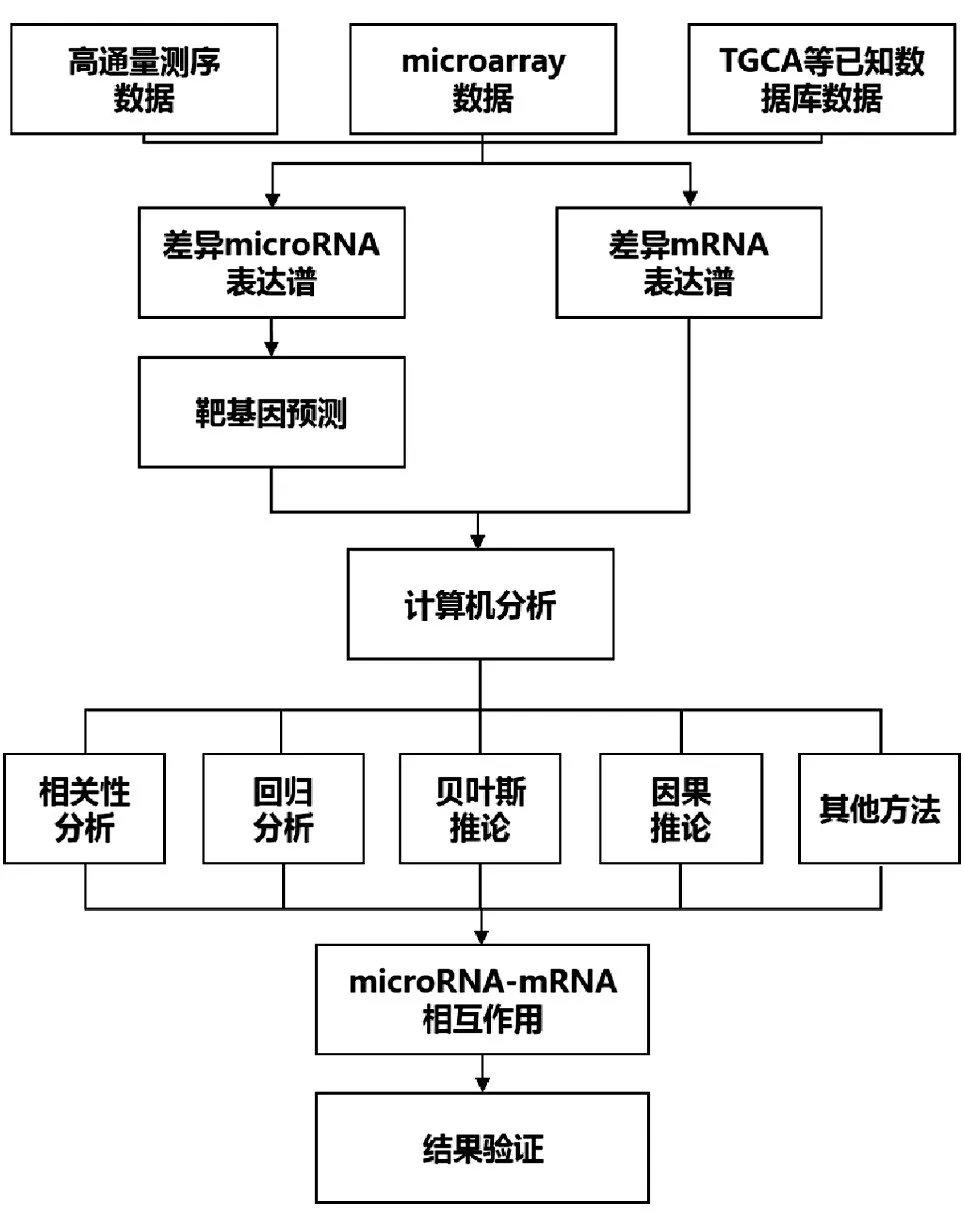
图1 MMIs分析流程图

表1 miRNA-mRNA常见分析方法
2.1 相关性分析
相关性分析是对2个或多个变量进行分析,以衡量变量间关系密切程度的一类统计方法。在MMIs中,miRNA主要对mRNA起负向调控作用(图2A),因此重点关注负相关系数的结果,其绝对值越大说明相关性越强。Pearson相关系数是最常用来反映2个正态分布的变量间线性关系的统计量[6]。在计算MMIs时,须提供5例以上样本的配对数据进行分析;当样本量较少(3~5例)时,可采用Spearman相关系数方法进行分析。Pear⁃son和Spearman方法的优势是计算简便、结果明确,但只能反映变量间的线性关系,而实际数据中变量间存在大量非线性关系,为此,有研究者提出使用互信息(mutual information,MI)和最大信息数(maximal information coefficient,MIC)等模型来衡量变量间的非线性强度。MI是信息论中的一种度量方法,关注2个随机变量之间的相互性,可以看成是一个随机变量中包含的关于另一个随机变量的信息量,适用于大样本量的分析[7],在MMIs分析中需要提供20例以上的配对表达谱(n>20)。MIC是在MI的基础上优化发展起来的一种分析方法,具有普适性、公平性、对称性等优势,当样本量足够时,可以捕获多种函数关系,被称为大数据时代相关性分析的最佳算法[8]。
2.2 回归分析
回归分析是判别因变量和自变量间关系的重要方法之一,它可以估计2个或2个以上变量间的关系,明确多个自变量对因变量的影响强度,构建预测模型。在实际MMIs中,1个miRNA可能调控多个靶基因,反之1个基因也可能被多个miRNA调控(图2B)。因此,单纯的相关性分析不能全面反映某种特定情况下miRNA-mRNA的实际关系,而回归分析则弥补了这种不足,更符合miRNA-mRNA调控的真实情况。Jayaswal等首次用最小二乘法回归对miRNA和mRNA表达值进行了MMI分析[9],随后Li等完善了部分算法并建立了结肠癌特异性的miRNA-mRNA调控网络[10]。近来,因为Lasso回归、Ridge回归和Elasticnet等3种分析方法能应对多重共线性数据,而被广泛应用于高通量数据相关性的预测中[11-13]。
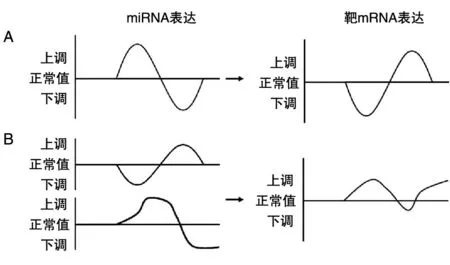
图2 不同分析方法预测miRNA-Target相关性的示意图
2.3 贝叶斯推理
传统的预测方法在预测过程中大多只利用模型和样本数据2种信息,而贝叶斯推理应用了决策者的主观信息,是对基于假设的先验概率进行修正的一种统计学方法。在MMIs分析中应用的预测假设是:①miRNA对mRNA起负调控作用;②当多个miRNA作用于同一个靶标时,对mRNA表达水平的影响是线性叠加。2007年,Huang等报道了一种运算法则——GenMiR++,他们用这种算法获得了一张包含104个miRNA和1597个靶标的网络图,并得出结论:与以序列为基础的预测相比,GenMiR++预测与基因注解有更高的一致性,是更精确的预测方法[14]。之后,他们在原有版本基础上完善了评估特异性序列(如高AU含量、杂交能量等)的功能,升级为GenMiR3模型[15]。由于GenMir++引入的不同推论的最大期望算法耗时很长,Su等引入了马尔可夫链蒙特卡洛算法,开发了HCtarget模型,增加了计算的时效性[16]。
2.4 因果推论
因果推论在相关性的基础上强调原因先于结果的特性,其应用于MMI分析的理论依据是:相关分析、回归分析和贝叶斯推理等研究方法关注miRNA-mRNA的相关性,而miRNA和mRNA之间的强相关性可能是由于mRNA对miRNA的调控作用或其他转录因子对二者共同的调控而产生的协同变化,不能说明二者之间的因果关系。经典的因果推论方法是在对照研究中通过敲除miRNA后观察靶mRNA表达发生的变化,但该方法耗时、耗力、耗钱。2000年Pearl等首次提出应用do-calculus方法对观测数据进行因果预测[17]。这种方法基于有向无环图(directed absent graph,DAG)的因果框架,但在实际计算中有时难以获得DAG,因此该方法不具有普适性。Maathuis等提出了一种名为IDA的方法,弥补了必须提供DAG结构的缺陷[18]。Le等认为miRNA与mRNA互作方式复杂,miRNA在调控mRNA的同时也会调控某些非编码RNA,包括其他miRNA。因此,他们在IDA的基础上增加了多对多的因果关系算法,并在miRNA敲除试验中得到了验证[19]。
2.5 其他方法
Z-score是用来预测基因敲除试验结果的一种网络方法,在敲除特定的miRNA后,可以计算出发生改变的基因偏离标准值的情况[20]。在分析MMI时,基于假设录入的miRNA的表达量为最低值,以此模拟miRNA被敲除的情况,再对应已知mRNA的表达量计算得分。Li等认为,尽管回归分析考虑了多个miRNA对同一个mRNA的竞争调控作用,但未考虑多个mRNA对同一个miRNA的竞争作用。当一个miRNA具有大量靶基因时,因为需要竞争靶结合位点,对每个靶标的调控作用将会被“稀释”。于是,他们开发了ProMISe模型,侧重预测mRNA与miRNA间的竞争性调节关系[21]。近来,Zoh等提出用典型相关分析方法很可能忽略二代测序中低表达(1~2个counts)数据间的强相关性,于是在Pearson相关系数和贝叶斯算法的基础上设计出PCAN模型,用于估算低表达量间的相关性[22]。
2.6 综合分析法
为了完整准确地预测miRNA-mRNA互作网络,Le等将多种方法组合后进行综合分析,采用波达计数法排列统计,最终提供研究者最为关注的Top k靶标列表(k值由研究者决定)。他们选用NCI-60数据库中的EMT(Epithelial to Mesen⁃chymal Transition)数据、MCC(Multi-class cancer)数据库中的多种癌症数据及BR51(51 human breast cancer cell lines)数据库中不同乳腺癌亚型的数据来综合比较不同综合分析方法的优缺点 ,利 用 Tarbase、miRecords、miRWalk 和 miRTar⁃Base等4个数据库交集的结果验证预测结果,最后得出Pearson+IDA+Lasso的组合是最优选的综合分析法的结论[23]。但由于综合分析法应用少,尚无足够证据证明综合分析法优于单一分析法。
3 MMIs分析常用软件
上述分析方法均可在相应的参考文献中找到对应的算法公式或开发的软件包供研究人员下载使用,以下重点对3个常用软件进行介绍。
MAGIA是由Bisognin等开发设计的一个MMI统计分析的网络软件,目前已升级到MAGIA2版本(http://gencomp.bio.unipd.it/magia2)[24-25]。使用者上传miRNA和对应的mRNA表达谱后,可以在8个miRNA靶标预测数据库(Microcosm、microrna.org、DIANA-microT、miRDB、PicTar、PITA、RNA22、TargetScan)中选择交集或并集的靶基因,再根据数据量、研究方向选择Spearman correlation、Pear⁃son correlation、MI(适用于20例以上的大样本)和Meta-analysis(适用于非配对样本)等4种统计方法中的一种进行关联分析,结果提供交互网络图和表格下载。MAGIA软件具有操作简单、使用方便、可直接提供调控网络图的输出等优点,但统计方法相对单一。
TaLasso是Muniategui等基于Lasso模型建立的网络分析软件(http://talasso.cnb.csic.es/)[26],需要研究者提供配对的miRNA和mRNA表达谱。在靶基因预测环节,提供 miRGen、miRBase、miRan⁃da、TarBase、miRecords和miRWalk等6个数据库可供选择。在联合分析时,除Lasso回归外,还可以选择GenMiR++或Pearson系数2种分析方法,结果提供得分和P值,并以表格形式输出。如果绘制网络图片,则需要使用Cytospace等绘图软件对结果进行二次制作。
miRLAB是Le等设计的一款基于R语言的软件包(http://bioconductor.org/packages/release/bioc/ht⁃ml/miRLAB.html)[27],下载安装后可在本地进行全部分析内容。该软件提供了3个基础数据库包,也可直接通过模块中TCGA数据库中的链接进行下载,或上传自备数据。获得原始数据后还可进行标准化、差异分析等预处理。在数据分析模块中,提供了12种MMI分析方式(Pearson相关、Spearman相关、Kendall等级相关、距离相关、Hoeffding相关、随机相关系数、MI、Lasso回归、Elastic-net回归、IDA、Z-score和 ProMISe)以供选择。在数据整合环节,在常规靶基因预测基础上增加了HITS-CLIP、PAR-CLIP和iCLIP的分析方式,可以直接揭示miRNA分子与其结合蛋白的相互作用。在结果验证环节,提供了既往验证的miRNA数据库和干扰试验。此外,还提供GO和KEGG等下游分析模块。与MAGIA和TaLasso相比,miRLAB的优势在于MMI分析方式多,使用范围广,但须掌握一定的R语言基础知识方能进行操作。
4 结语
目前人们对miRNA的研究仍处于早期探索阶段,大部分研究还局限于通过生物信息学软件结合miRNA的部分特征对其靶标和功能进行预测。但这种方法假阳性率高,不具有普遍适用性,预测效果并不乐观。随着高通量技术的广泛应用,利用miRNA和mRNA表达谱进行联合分析越来越受到人们的认可并逐渐普及。尽管现有模型预测的结果仍然不够准确,且需要下游实验验证,但随着对miRNA作用机制的深入研究,基于配对表达谱进行联合预测的方法也在日渐丰富和完善。相信在不久的将来,在多学科研究人员的共同努力下,MMIs将会出现更加便捷、全面、准确的研究方法。
[1] Rana T M.Illuminating the silence:understanding the structure and function of small RNAs[J].Nat Rev Mol Cell Biol,2007,8(1):23-36.
[2] Hobert O.Gene regulation by transcription factors and miRNAs[J].Science,2008,319(5871):1785-1786.
[3] Enright A J,John B,Gaul U,et al.microRNA tar⁃gets in Drosophila[J].Genome Biol,2003,5(1):R1.
[4] Rajewsky N.miRNA target predictions in animals[J].Nat Genet,2006,38(Suppl):S8-13.
[5] Farazi T A,Spitzer J I,Morozov P,et al.miRNAs in human cancer[J].J Pathol,2011,223(2):102-115.
[6] Speed T.Mathematics.A correlation for the 21st cen⁃tury[J].Science,2011,334(6062):1502-1503.
[7] Moon Y I,Rajagopalan B,Lall U.Estimation of mutu⁃al information using kernel density estimators[J].Phys Rev E Stat Phys Plasmas Fluids Relat Interdiscip Top⁃ics,1995,52(3):2318-21.
[8] Reshef D N,Reshef Y A,Finucane H K,et al.De⁃tectingnovel associationsin largedatasets[J].Sci⁃ence,2011,334(6062):1518-1524.
[9] Jayaswal V,Lutherborrow M,Ma D D,et al.Identifi⁃cation ofmiRNAswith regulatorypotentialusinga matched miRNA-mRNA time-course data[J].Nucleic Acids Res,2009,37(8):e60.
[10]Li X,Gill R,Cooper N G,et al.Modeling miRNA-mRNA interactions using PLS regression in human co⁃lon cancer[J].BMC Med Genomics,2011,4:44.
[11]Ragan C,Zuker M,Ragan M A.Quantitative predic⁃tion of miRNA-mRNA interaction based on equilibri⁃um concentrations[J].PLoS ComputBiol,2011,7(2):e1001090.
[12]Nogales-Cadenas R,Carmona-Saez P,Vazquez M,et al.GeneCodis:interpreting gene lists through enrich⁃ment analysis and integration of diverse biological in⁃formation[J].Nucleic Acids Res,2009,37(Web Server issue):W317-322.
[13]Muniategui A,Pey J,Planes F J,et al.Joint analysis of miRNA and mRNA expression data[J].Brief Bioin⁃form,2013,14(3):263-278.
[14]Huang J C,Babak T,Corson T W,et al.Using ex⁃pression profiling data to identify human miRNA tar⁃gets[J].Nat Methods,2007,4(12):1045-1049.
[15]Huang J C,Frey B J,Morris Q D.Comparing se⁃quence and expression for predicting miRNA targets using GenMiR3[C]//Proceedings of the Pacific Sympo⁃sium.Biocomputing 2008.2008:52-63.
[16]Su N Wang Y,Qian M,et al.Predicting microRNA targets by integrating sequence and expression data in cancer[JC]//IEEE Int Conf Syst Biol.2011:219-224.
[17]PearlJ.Causality:models,reasoning,and inference[M].Cambridge University Press,2000:384.
[18]Maathuis M H,Colombo D,Kalisch M,et al.Predict⁃ing causal effects in large-scale systems from observa⁃tional data[J].Nat Methods,2010,7(4):247-248.
[19]Le T D,Liu L,Tsykin A,et al.Inferring miRNA-mRNA causal regulatory relationships from expression data[J].Bioinformatics,2013,29(6):765-771.
[20]Prill R J,Marbach D,Saez-Rodriguez J,et al.To⁃wards a rigorous assessment of systems biology mod⁃els:the DREAM3 challenges[J].PLoS One,2010,5(2):e9202.
[21]Li Y,Liang C,Wong K C,et al.Inferring probabilis⁃tic miRNA-mRNA interaction signatures in cancers:a role-switch approach[J].Nucleic Acids Res,2014,42(9):e76.
[22]Zoh R S,Mallick B,Ivanov I,et al.PCAN:probabilis⁃tic correlation analysis of two non-normal data sets[J].Biometrics,2016,72(4):1358-1368.
[23]Le T D,Zhang J,Liu L,et al.Ensemble methods for miRNA target prediction from expression data[J].PLoS One,2015,10(6):e0131627.
[24]Bisognin A,Sales G,Coppe A,et al.MAGIA(2):from miRNA and genes expression data integrative analysis to miRNA-transcription factor mixed regulatory circuits[J].Nucleic AcidsRes,2012,40(Web Serverissue):W13-21.
[25]Sales G,Coppe A,Bisognin A,et al.MAGIA,a webbased tool for miRNA and genes integrated analysis[J].Nucleic AcidsRes,2010,38(Web Serverissue):W352-359.
[26]Muniategui A,Nogales-Cadenas R,Vazquez M,et al.Quantification of miRNA-mRNA interactions[J].PLoS One,2012,7(2):e30766.
[27]Le T D,Zhang J,Liu L,et al.miRLAB:an R based dry lab for exploring miRNA-mRNA regulatory rela⁃tionships[J].PLoS One,2015,10(12):e0145386.
Progressin the Integrative AnalysisofmicroRNA and mRNA Expression Data
ZHAO Lu-Yang1a,ZHANG Kang2,GU Cheng-Lei1a,3,YE Ming-Xia1a,FAN Wen-Sheng1a,HAN Wei-Dong1b,MENG Yuan-Guang1a*
1.a.Department of Gynecology and Obstetrics;b.Institute of Basic Medicine;Chinese PLA General Hopsital,Bei⁃jing 100853;2.Department of Gynecology and Obstetrics,Beijing Huaxin Hospital,Beijing 10016;3.Department of Gynecology and Obstetrics,PLA 309 Hospital,Beijing 100091;China
*Corresponding author,E-mail:meng6512@vip.sina.com
microRNAs(miRNAs) are endogenous non-coding small RNAs that interact with their mRNAs by de⁃grading or inhibiting translation of the targets.Up to date,there is still no low-cost and effective miRNA target screening method because the regulatory mechanisms are complex.In recent years,some investigators built several computational methods based on sequence complementarity of the miRNA and the mRNAs.However,the results are various out of different algorithms and have huge rate of false positives,which caused great troubles in down⁃stream experiments.Therefore,methods of using the expression values of miRNAs and mRNAs to refine the re⁃sults has been proposed,which have shown to effectively identifying the most prominent interactions.Here,we summarized these methods that combining both miRNA expression values and mRNA expression values and to pre⁃dict miRNA targets,outlined the advantages and disadvantages of different methods,and provided directions for fu⁃ture investigation.
microRNA;microRNA target prediction;microRNA-mRNA integration;integrative analysis
Q78;Q811.4
A
1009-0002(2017)04-0545-06
2016-12-22
国家自然科学基金面上项目(81571411)
赵路阳(1988- ),女,博士研究生,(E-mail)zlynfyd@163.com
孟元光,(E-mail)meng6512@vip.sina.com
10.3969/j.issn.1009-0002.2017.04.029
猜你喜欢
北京航空航天大学学报(2021年9期)2021-11-02 08:24:32
军民两用技术与产品(2021年10期)2021-03-16 06:05:10
重型机械(2020年2期)2020-07-24 08:16:16
学苑创造·A版(2020年12期)2020-01-07 14:07:23
中国外汇(2019年15期)2019-10-14 01:00:34
世界农药(2019年3期)2019-09-10 07:04:10
中国航海(2019年2期)2019-07-24 08:26:40
光学精密工程(2016年4期)2016-11-07 09:04:48
作文教学研究(2016年1期)2016-07-05 12:22:47
肿瘤影像学(2015年3期)2015-12-09 02:38:45
